
Abstract
In light of the most challenging public health crisis of modern history, COVID-19 mortality continues to rise at an alarming rate. Patients with co-morbidities such as hypertension, cardiovascular disease, and diabetes mellitus (DM) seem to be more prone to severe symptoms and appear to have a higher mortality rate. In this review, we elucidate suggested mechanisms underlying the increased susceptibility of patients with diabetes to infection with SARS-CoV-2 with a more severe COVID-19 disease. The worsened prognosis of COVID-19 patients with DM can be attributed to a facilitated viral uptake assisted by the host’s receptor angiotensin-converting enzyme 2 (ACE2). It can also be associated with a higher basal level of pro-inflammatory cytokines present in patients with diabetes, which enables a hyperinflammatory “cytokine storm” in response to the virus. This review also suggests a link between elevated levels of IL-6 and AMPK/mTOR signaling pathway and their role in exacerbating diabetes-induced complications and insulin resistance. If further studied, these findings could help identify novel therapeutic intervention strategies for patients with diabetes comorbid with COVID-19.
Similar content being viewed by others


1 Introduction
Over the last two decades, severe acute respiratory infection outbreaks have accompanied a generalized global health concern. Two prominent coronaviruses, severe acute respiratory syndrome coronavirus (SARS-CoV-1) and the Middle East respiratory syndrome coronavirus (MERS-CoV), have been associated with a high pathogenicity and mortality in humans [1]. SARS-CoV-1, which emerged from 2002 to 2003, caused over 8000 confirmed cases of infection and about 800 deaths, while MERS-CoV, which was first reported in 2012, is still present to date and has infected over 2300 individuals worldwide [1, 2]. Yet, these two coronaviruses never reached a level of pandemic.
In December of 2019, a series of pneumonia cases with unknown etiology were reported in Wuhan, a city in the Hubei province of China. High-throughput sequencing from lower respiratory tract samples revealed a novel coronavirus named 2019 novel coronavirus (2019-nCoV). However, as suggested by a recent study based on validated satellite imagery data of hospital parking lots and Baidu search queries of disease related terms, the virus may have already been circulating when the outbreak was declared. This recent evidence shows an upward trend in hospital traffic and search volume beginning in late Summer and early Fall 2019 as well as an increase in searching for the terms ‘cough’ and ‘diarrhea’, the latter being a more specific symptom for COVID-19 [3]. The increasing number of cases urged the World Health Organization (WHO) to declare a Public Health Emergency of International Concern on January 30, 2020. The novel virus was then formally referred to as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the disease as coronavirus disease 2019 (COVID-19). As testing became more available, the number of new cases increased exponentially, and on March 11, 2020, the WHO declared the outbreak a global pandemic [4].
Ever since its discovery in late 2019, SARS-CoV-2 has quickly spread to more than 200 countries around the world. As of June 28, 2020, more than 10 million cases of COVID-19 have been reported with a death toll of 501,469 individuals. Despite the low mortality rate of COVID-19, patients with co-morbidities such as hypertension, cardiovascular disease, and diabetes mellitus seem to be prone to more severe symptoms and to a higher mortality rate than others [5, 6]. Obesity also appears to worsen the prognosis of patients with COVID-19, specifically in younger obese individuals who seem to also be susceptible to a more severe disease [7]. Increasing evidence highlight diabetes mellitus as a distinct comorbidity associated with acute respiratory distress syndrome (ARDS) and increased subsequent mortality [6, 8, 9].
In the realm of social distancing imposed by the pandemic, the health care management system was found to be overwhelmed by the rapidly increasing demand on health facilities. The lack of previous preparedness was further challenged by fears of an imminent worldwide economic crisis that accelerated the race to understand the pathogenesis of SARS-CoV-2 in order to develop novel therapeutic strategies.
COVID-19 clinical signs are diverse, ranging from an asymptomatic state to ARDS and multi-organ dysfunction [10], with respiratory failure from ARDS being the leading cause of mortality [11]. COVID-19 symptoms generally manifest after an estimated incubation period of approximately one week (mean = 7 days, range = 0–24 days) [6]. The most common clinical features include fever, cough, and fatigue, while other symptoms include headache, hemoptysis, diarrhea, dyspnea, and lymphopenia [4, 12,13,14,15]. In a subset of patients, the disease rapidly progressed to severe chest pain, pneumonia and ARDS by the end of the first week [4]. In severe cases, SARS-CoV-2 virus targets both the upper and lower respiratory tract, causing irreversible injuries, notably pulmonary fibrosis [8, 16, 17]. COVID-19-associated pulmonary complications are exacerbated in patients with co-morbidities such as hypertension, cardiovascular disease, obesity and diabetes mellitus [5,6,7].
In this review, we detail our present understanding of the pathogenesis of SARS-CoV-2 and elucidate possible mechanisms behind the increased susceptibility of patients with diabetes to infection with more serious complications.
2 COVID-19 pathogenesis
2.1 The immune response to SARS-CoV-2
COVID-19 belongs to the coronavirus family, a large family of single-stranded enveloped RNA viruses that is divided into four genera: Alpha-, Beta-, Delta- and Gammacoronavirus [18]. Coronaviruses from the genera Alpha- and Betacoronavirus are primarily associated with infections in mammals, while viruses in the genera Gamma- and Deltacoronavirus mainly infect birds [18]. Both SARS-CoV-1, the virus responsible for the 2002 outbreak, and SARS-CoV-2, belong to the β-genus [19]. Many of the symptoms caused by SARS-CoV-2, such as ARDS, are quite similar to those resulting from SARS-CoV-1 [15]. These similarities can be traced back to the structural analogy between the two virus’ envelope-anchored spike (S) protein, which mediates their entry into the host cells [20]. Extensive studies on SARS-CoV-1 have identified key interactions between its S protein receptor-binding domain (RBD) and its host receptor angiotensin-converting enzyme 2 (ACE2), which control its cross-species and human-to-human transmissions [20, 21]. SARS-CoV-1 and SARS-CoV-2’s respective S proteins share a 76% to 78% sequence analogy for the whole protein and a 73% to 76% sequence similarity for the RBD, strongly suggesting that both viruses share the same access door to host cells: the angiotensin-converting enzyme 2 (ACE2) [19] (Fig. 1).

Schematic diagram representing (a) SARS-CoV-2 entry into the host cell and (b) the role of ACE2 in the renin-angiotensin system
Besides, SARS-CoV-2 infection leads to an increased release of pro-inflammatory cytokines and chemokines including interleukins IL-1β, IL-4, and IL-10, monocyte chemoattractant protein 1 (MCP-1), interferon- γ (IFNγ), and interferon gamma-induced protein 10 (IP-10) [15]. Notably, ICU patients with severe disease had significantly elevated plasma levels of IL-2, IL-6, IL-7, IL-10, granulocytes colony stimulating factor (GCSF), IP-10, MCP-1, macrophage inflammatory protein-1A (MIP-1A), and tumor necrosis factor-α (TNF-α), suggesting a potential “cytokine storm” correlated with COVID-19 disease severity [15, 22]. The release of pro-inflammatory cytokines and chemokines may potentially be attributed to massive epithelial and endothelial cell apoptosis and to vascular leakage resulting from rapid viral replication [23]. Of the released pro-inflammatory cytokines, IL-1β and IL-6 are of particular interest and appear to be closely related to the occurrence of severe COVID-19 in adult patients [24]. This hypercytokinemia seems to play a crucial role in the development of pulmonary fibrosis [25] and is associated with increased viral load, loss of lung function, lung injury, and increased mortality [26].
IL-1β was shown to be increased in the bronchoalveolar lavage fluid and in the plasma of patients with ARDS [27]. Similarly, IL-6 functions as a proinflammatory factor and was shown to play an important role in the progression of lung fibrosis [28]. IL-6 is an important pleiotropic cytokine that significantly contributes to acute inflammation. Elevated IL-6 levels were correlated with increased severity of COVID-19-associated pneumonia. In mild cases, systemic levels of IL-6 were less than 100 pg/mL. However, in critical cases, IL-6 levels were greater than 100 pg/mL, a concentration above which we usually witness the emergence of an “inflammatory storm” [29]. Consequently, it was reported that the inhibition of both IL-1β and IL-6 is beneficial in many viral infections [24]. A retrospective study observing the efficacy of tocilizumab (IL-6R antagonist) in treating COVID-19 suggested that tocilizumab might be an effective treatment in patients with the severe form of the disease [30]. Currently, several clinical trials on the safety and efficacy of tocilizumab in the treatment of severe COVID-19-associated pneumonia in adult inpatients have been registered [31, 32]. Interestingly, IL-6 activation is directly correlated with the Mechanistic Target of Rapamycin (mTOR) pathway activation, a pathway involved in cell survival, proliferation, and growth [33]. In that spirit, cytokine IL-37, which has the ability to suppress both the innate and the acquired immune responses and to inhibit inflammation by acting on IL-18Rα receptor, was also shown to suppress the production of IL-1β and IL-6 by modulating mTOR pathway and increasing the adenosine monophosphate kinase (AMPK) [24]. IL-38 is another inhibitory cytokine of IL-1β and other pro-inflammatory IL-family members [24]. Both IL-38 and IL-37 were suggested to serve as potential therapeutic cytokines by inhibiting inflammation caused by COVID-19, providing a novel pertinent approach to treating the disease.
2.2 The protective role of angiotensin-converting enzyme 2 against lung injury
The pathophysiology of SARS-CoV-2 infection has not yet been extensively investigated, but it is speculated that it can resemble that of SARS-CoV-1 overall. Infection with SARS-CoV-1 results in an aggressive inflammatory response that begins with binding to the membrane-bound ACE2 receptor [19] followed by entry into the cell and subsequent viral replication [34]. Similarly, a possible mechanism of SARS-CoV-2-mediated inflammatory responses consists of downregulation and shedding of ACE2, a terminal carboxypeptidase that degrades angiotensin II to angiotensin (1–7), thus acting as a negative regulator of the renin-angiotensin system [35]. While ACE, which converts angiotensin I to angiotensin II, induces lung edema and promotes lung injury, ACE2 appears to protect the lungs from acute injury [36] (Fig. 1). In several studies, loss of pulmonary ACE2 expression resulted in increased inflammation, enhanced vascular permeability, increased lung edema, and accumulation of neutrophils, eventually leading to decreased lung function [35,36,37,38]. Previous studies on SARS-CoV-1 have shown that once bound to ACE2 the virus’ S protein downregulates ACE2 [39, 40] and leads to the shedding of its ectodomain, an enzymatically active domain termed soluble ACE2 (sACE2) [41,42,43]. The biological function of sACE2 remains poorly investigated. Inflammatory cytokines such as IL-1β and TNF-α were also shown to increase ACE2 shedding [41,42,43]. Thus, for SARS-CoV-2 pathogenesis, ACE2 not only serves as a portal entry for the virus but also plays a protective role against lung injury. These observations hypothesize that increased ACE2 shedding, which is correlated with the uncontrolled inflammation in SARS-CoV-1 infection, might also be involved in the hyperinflammation seen with SARS-CoV-2 infection.
After reviewing the epidemiology and pathogenesis of SARS-CoV-2, it is well recognized that DM increases morbidity and mortality in patients with COVID-19 by aggravating the pathogenesis of the disease [6, 8, 9, 44, 45]. Herein, we aim to elucidate the pathological mechanisms in relation to diabetes and COVID-19 and suggest potential therapeutic strategies for managing the complications emanating from the viral infection.
3 Diabetes mellitus: A risk factor for the progression of COVID-19
Diabetes mellitus is one of the leading causes of morbidity worldwide, and it is projected to remain on the rise over the next few decades. A large body of evidence has highlighted an increased susceptibility of patients with diabetes to infectious diseases [46,47,48], which is possibly attributed to a defective immune system in diabetes [49]. Given the decreased immunity in patients with diabetes, pneumonia has now become a considerable mortality factor in diabetes [50]. In patients with SARS, diabetes and plasma glucose levels were both shown to be independently associated with higher morbidity and mortality [51]. In Hong Kong, the first three deaths from SARS-CoV-2 infection were patients with diabetes. In a study conducted on a group of 52 ICU patients infected with SARS-CoV-2, the most common comorbidities between the 32 non-survivors of the group were diabetes (22%) and cerebrovascular disease (22%) [8]. Recently, The Chinese Center for Disease Control and Prevention published the largest study relevant to patients with diabetes in Mainland China which involved 72,314 cases of COVID-19. While patients who reported no co-morbidities had a case fatality rate of 0.9%, patients with diabetes had a significantly higher case fatality rate (7.3%) [52]. Furthermore, a meta-analysis of 76,993 patients infected with SARS-CoV-2 revealed that hypertension, cardiovascular disease, history of smoking, and diabetes were the most common underlying diseases with incidences of 16.37%, 12.11%, 7.63%, and 7.87%, respectively [53]. In another study conducted on 1099 COVID-19-infected patients in China, 173 cases (16%) were classified as severe [6]. Out of these severe cases, 16.2% (28 individuals) had diabetes, while only 5.7% (81 individuals) of the non-severe cases had diabetes [6]. Furthermore, a retrospective study in Wuhan, China conducted on 174 patients with COVID-19 revealed a higher risk of severe pneumonia in patients with diabetes (n = 24) who did not suffer from any other complication [54]. These patients also presented with a higher risk of tissue injury-related enzyme release and an overexpressed uncontrolled inflammation. Dysregulated glycemia also appeared to lead to a hypercoagulable state through the activation of plasmin, thrombin and monocytes-macrophages and through the secretion of different tissue factors, a resultant of the inflammatory storm itself [54]. According to the CDC, as of May 30, 2020, in a population of about 1.3 million individuals infected with SARS-CoV-2 in the USA, around 30% of those individuals who have underlying health conditions (86,737 individuals) have diabetes mellitus [55]. The different studies presented suggest that patients with diabetes may not only be prone to a more severe COVID-19 disease, but also to an increased risk of infection with SARS-CoV-2. However, several studies have shown that, despite these latter findings, no increased infectivity was observed in patients with COVID-19 comorbid with diabetes [56]. In fact, the prevalence of diabetes in the patient population with COVID-19 is not so different from the prevalence of diabetes in the general population [56].
4 Elucidating the crosstalk between COVID-19 and diabetes mellitus
Several mechanisms were suggested to explain the increased susceptibility of patients with DM to severe COVID-19 disease, including higher-affinity cellular binding, efficient viral entry, reduced viral clearance, reduced T cell function, enhanced susceptibility to hyperinflammation and cytokine storm, and the presence of cardiovascular diseases [57]. Phagocytosis by neutrophils, monocytes, and macrophages was shown to be defective in patients with diabetes who happen to also suffer from malfunctions in neutrophil chemotaxis, bactericidal activity, and innate cell-mediated immunity [58]. Interestingly, even short-term hyperglycemia was found to dampen their innate immune response [59]. In addition to their defective innate response, patients with diabetes also demonstrate an impaired adaptive immune response [49].
4.1 Angiotensin-converting enzyme 2 expression in diabetes mellitus and its role in COVID-19 infectivity
Although plausible hypotheses for the increased risk of COVID-19 infection in patients with diabetes and other chronic diseases like hypertension are still under investigation, ACE2 seems to play a key role in the association between COVID-19 and DM [60] (Table 1). In fact, both DM and hypertension are correlated with the activation of the renin-angiotensin system in different tissues [71], a system that regulates blood volume and the systemic vascular resistance [72]. Treating type 1 and type 2 diabetes with ACE inhibitors and angiotensin II type-I receptor blockers (ARBs) was found to increase the expression of ACE2 in the renal and cardiovascular systems [61, 62]. However, there is no sufficient evidence to support an increase in ACE2 levels in the respiratory system secondary to the use of ACE/ARBs.
There is also no sufficient experimental evidence to support the hypothesis that switching people from ACE inhibitors or ARBs to other drugs might decrease the risk of infection and the severity of COVID-19. A more favorable SARS-CoV-2 binding was demonstrated with increased ACE2 expression in alveolar AT2 cells, as well as in the myocardium, kidneys, and pancreas in humans [63,64,65]. In rodents with DM, an increased expression of ACE2 was also reported in the lungs, kidneys, heart, and pancreas [66, 67], thus possibly favoring SARS-CoV-2 entry. Hypoglycemic agents such as thiazolidinediones (TZDs; pioglitazone) and glucagon-like peptide–1 (GLP-1) agonists (liraglutide), statins and ibuprofen were all also found to increase ACE2 expression [73,71,72,76]. This explains why concerns were initially raised regarding the use of non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen in patients with COVID-19. The WHO later denied this assumption, stating that no severe adverse effects were observed with the use of NSAIDs in patients with COVID-19 [77]. Until recently, the association between DM and ACE2 expression levels in human lungs remained poorly investigated. A phenome-wide Mendelian randomization analysis carried out by Rao et al. suggested that higher ACE2 expression in the lungs increased susceptibility to SARS-CoV-2 infection with more severe complications and was causally correlated with diabetes [68]. The genome-wide association study (GWAS) on patients with type 2 diabetes (N = 898,130) revealed that type 2 diabetes is causally linked to increased ACE2 expression. Another study showed that patients with diabetes have increased levels of furin [78], a cellular protease that cleaves the S1 and S2 domains of SARS-CoV-2’s spike protein [79], possibly facilitating viral entry.
The role of ACE2 in the crosstalk between COVID-19 and DM is still a matter of debate. Some studies recognized decreased levels of ACE2 in diabetes, perhaps secondary to glycosylation [80]. In kidney biopsies of patients with diabetes presenting with nephropathy, glomerular expression of ACE2 was also found to be reduced [69]. Therefore, by adopting the hypothesis that increased ACE2 expression leads to higher viral infectivity, it would be reasonable to infer that diabetes, with its diminished ACE2 expression, is associated with a lower risk of infection with SARS-CoV-2. However, as previously mentioned, diabetes was shown to be associated with a higher risk of severe COVID-19 disease and a poorer prognosis. This highlights the presence of other factors that explain the positive association between diabetes and COVID-19. Treatment with ACE inhibitors, ARBs, TZD or GLP-1, higher levels of furin, delayed viral clearance, immune dysfunction, comorbidities, and other confounding factors could all explain the higher prevalence of COVID-19 in patients with diabetes.
Remarkably, other studies argue that having higher ACE2 expression does not lead to increased infectivity and severity of COVID-19 disease. On the contrary, some consider that increased ACE2 could play a beneficial role in patients with COVID-19 [81, 82]. Patients with diabetes treated with ACE inhibitors or ARBs might thus be at an advantage over non-treated patients with diabetes [81, 82]. As previously mentioned, loss of pulmonary ACE2 expression leads to decreased lung function. This justifies why treatment with ACE inhibitor or ARBs has been advanced as a possible therapeutic strategy for COVID-19 [81, 82]. Moreover, it has been lately proposed that treatment with a soluble form of ACE2, which lacks the membrane anchor, may act as a competitive interceptor of SARS-CoV-2 by inhibiting the binding of the virus’ S protein to the surface-bound, full-length ACE2 [83]. A recent study has shown that treatment with clinical-grade human recombinant soluble ACE2 (hrsACE2) significantly inhibited viral growth in the monkey kidney cell line, Vero-E6, by a factor of 1000-5000. It also prevented SARS-CoV-2 infection in engineered human blood vessel organoids and human kidney organoids at the early stage of infection [70]. Collectively, these findings highlight the need for a better understanding of the underlying pathobiology of ACE2 in DM and COVID-19.
Based on these studies, the interplay between diabetes and COVID-19 appears to be bi-directional. Diabetes was shown to be associated with an increased risk of severe COVID-19. New-onset diabetes and severe metabolic complications of preexisting diabetes, such as diabetic ketoacidosis and hyperosmolarity, were also noted in patients with COVID-19, which was proposed to be due to the binding of SARS-CoV-2 to ACE2 receptors in key metabolic organs, possibly resulting in variations in glucose metabolism [84,85,86,87]. ACE2 expression is particularly amplified in key metabolic organs such as the liver, the endocrine pancreas, adipose tissue, the kidneys and the small intestine, which might play a role in the emergence of insulin resistance, as well as in the impaired secretion of insulin [88, 89]. Thus, it could be hypothesized that SARS-CoV-2 infects metabolic organs, leading to hyperglycemia exacerbation. More importantly, a subclinical inflammatory reaction, in particular a combined elevation of IL-1β and IL-6, has been shown to precede the onset of type 2 diabetes [90], further suggesting that COVID-19 might increase the risk of developing new-onset diabetes.
4.2 Diabetes mellitus and COVID-19: In the eye of the “cytokine storm”
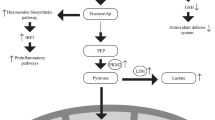
Another potential reason for the increased risk of severe COVID-19 disease in patients with diabetes might be attributed to the hyperinflammatory response, referred to as “cytokine storm” (Fig. 2). Patients with diabetes suffer from a continuous low-grade inflammation facilitating the emergence of a cytokine storm, which in turn appears to be directly related to the severity of COVID-19 pneumonia cases and to subsequent death [91]. Patients with diabetes appear to have an impaired adaptive immune response characterized by an initial delay of Th1 cell-mediated immunity and a late hyperinflammatory response [49]. In the absence of an immunostimulant, diabetes is associated with an increased pro-inflammatory cytokine response marked by increased secretion of IL-1, IL-6, IL-8 and TNF-α [58]. Elevated basal cytokine levels might also be attributed to advanced glycation end products (AGEs) [92], which consist of residues of glucose and lysine/arginine [58]. It was noted that extended emergence of AGEs occurred in poorly regulated patients with diabetes. Separate studies have established an increase in cytokine levels following AGE binding to non-diabetic cells, without direct stimulation [93,94,95]. As such, elevated AGE production in patients with diabetes could be implicated in raising resting cytokine production [58]. Other studies evaluated the responsiveness of peripheral blood mononuclear cells (PBMCs) and isolated monocytes in patients with diabetes after being subjected to stimulation. Interestingly, IL-1 and IL-6 secretion resulting from exposure to lipopolysaccharide (LPS) was found to be diminished in patients with diabetes [58, 96, 97]. One might speculate that the high resting value of diabetic cells could favor tolerance to direct stimulation, with an ensuing decrease in the cytokine secretion response. This type of event has already been reported in non-diabetic cells [98]. In short, the mere availability of high glucose causes an increase in resting cytokine production; yet, subsequent to stimulation, cytokine production lessens in comparison to a condition without glucose. This reduction in interleukin production upon stimulation might also be attributed to intrinsic cellular defects in patients with diabetes [58, 99].

COVID-19, Diabetes Mellitus, cytokine storm: a vicious cycle
Chronic inflammation reported in DM is further amplified with SARS-CoV-2 infection, resulting in an aggressive inflammatory response. A study done on a humanized mouse model of MERS-CoV infection revealed that the disease was more severe and prolonged in male diabetic mice and was characterized by alterations in CD4+ T cell counts and abnormal cytokine responses [100]. In accordance with this animal study, other studies conducted on patients with diabetes comorbid with COVID-19 have observed decreased peripheral CD4+ and CD8+ T cells counts and increased cytokine levels [6, 8, 9, 45, 101]. A recent study revealed that patients with diabetes comorbid with COVID-19, despite having significantly lower absolute lymphocyte counts in peripheral blood, had notably higher absolute neutrophil counts in comparison to non-diabetic patients [54]. Among the different markers of inflammation found to be elevated in COVID-19 cases with diabetes, IL-6 warrants particular attention since it has been shown to be associated with lung injury and poorer prognosis [28, 102]. Interestingly, serum levels of IL-6 in diabetic patients without COVID-19 were significantly higher compared to those in non-diabetic patients [54]. This might be correlated with the increased cytokine baseline level observed in DM, which is further amplified in COVID-19. These findings indicate that IL-6 might be a good predictor of disease severity and prognosis. They also further suggest that patients with both diabetes and COVID-19 are susceptible to a more aggressive inflammatory storm, ultimately leading to rapid deterioration.
Taken together, these observations suggest that tocilizumab (IL-6R antagonist) may markedly help in the treatment of COVID-19 pneumonia. In fact, tocilizumab is now being used off-label in some Italian centers in patients with COVID-19 and is currently being assessed in an ad hoc randomized controlled trial [31]. Moreover, IL-37, which decreases the production of IL-1β and IL-6 by modulating the mTOR pathway and increasing AMPK, was suggested as a potential treatment for COVID-19 [24]. Of note, it was previously shown that kinase inhibitors targeting PI3K/AKT/mTOR pathways also significantly inhibited MERS-CoV replication in vitro [103]. Interestingly, AMPK/mTOR signaling pathway is well known to be altered in DM. This not only provides a plausible explanation for the increased susceptibility of patients with diabetes to COVID-19, but also alludes to the possible role of COVID-19 in worsening diabetes and diabetes-induced complications. Yet, the role of AMPK/mTOR signaling axis in COVID-19 associated complications should be further studied.
4.3 The interplay between COVID-19 and AMPK/mTOR signaling pathway in diabetes mellitus
AMPK is a key physiological energy sensor whose activity is regulated by glucose. AMPK signaling modulates multiple biological pathways such as cellular metabolism, growth and proliferation to maintain cellular energy homeostasis [104]. A major downstream signaling pathway regulated by AMPK is the mTOR pathway. mTOR is a serine/threonine protein kinase that exists in 2 complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The two subtypes consist of distinct sets of protein-binding partners [105]. mTORC1 comprises mTOR, mLST8 and rapamycin-sensitive adaptor protein of mTOR (Raptor) and is known to mediate many of its downstream effects including protein synthesis and cell size through p70S6 kinase (p70S6K)/S6 kinase 1 (S6K1) and 4E-binding protein 1 (4E-BP1) [106,107,108]. mTORC2, with its essential components mTOR, mSIN1, mLST8, and the rapamycin-insensitive subunit Rictor, mediates its actions through the phosphorylation of protein kinase B (PKB/Akt) at Serine 473 [109]. mTORC2 has been implicated in controlling cell survival and cytoskeletal organization [109]. When cellular energy levels are low, AMPK is activated to stimulate glucose uptake in skeletal muscles and fatty acid oxidation in adipose tissues. It also reduces hepatic glucose production. A large body of evidence underlines a dysregulation in AMPK signaling in metabolic syndrome and DM [110,111,112,113]. More importantly, it has been previously shown that AMPK activation can improve insulin sensitivity by enhancing glucose transport and uptake and by stimulating fatty acid oxidation [114]. In 2001, metformin was reported to act as an AMPK activator and is now a widely used drug for the treatment of type 2 diabetes [115], and recently for type 1 diabetes [116]. Strong evidence demonstrates that AMPK negatively regulates the mTOR pathway. It inhibits mTORC1 indirectly through the phosphorylation of Tuberous sclerosis 2 (TSC2), thus favoring a TSC1-TSC2 association, an upstream inhibitor complex of mTORC1. Furthermore, AMPK modulates mTORC1, independently from TSC2 by raptor phosphorylation and inactivation of mTORC1 [117].
During the progression of DM, AMPK is inactivated leading to chronic overactivation of mTORC1 [118,119,120]. Overactivation of mTOR signaling pathway has been associated with insulin resistance and progression of diabetes-induced complications. For instance, our group has previously shown that hyperglycemia was associated with increased activation of mTORC1/p70 S6Kinase and Rictor/mTORC2 pathways through the inactivation of AMPK, eventually leading to podocyte injury. Intriguingly, inhibition of mTORC1 by rapamycin or of mTORC2 by using antisense oligonucleotides that target Rictor attenuated glomerular injury and prevented podocyte loss/depletion [110, 112].
Although not well elucidated, the effects of metformin are thought to be mediated by the regulation of AMPK and mTOR. Intriguingly, a recent study has shown that B cell function and influenza vaccine responses attenuated by type 2 diabetes and obesity were improved by metformin [121]. Moreover, metformin decreased B cell intrinsic inflammation and increased antibody responses when used in vitro to stimulate B cells isolated from patients with recently diagnosed type 2 diabetes [121]. These findings suggest that metformin activates AMPK consequently leading to an improvement in B cell responses and a decrease in B cell intrinsic inflammation. Therefore, AMPK is proposed as a potential therapeutic target in viral infections.
Increasing evidence also highlight mTORC1 as a key player in controlling the replication of viruses such as Andes orthohantavirus and coronavirus [122, 123]. In patients with H1N1 pneumonia and acute respiratory failure, treatment with corticosteroids and an mTOR inhibitor effectively blocked viral protein expression and virion release, attenuated hypoxia and multiorgan dysfunction and improved patients’ prognosis significantly [124]. Furthermore, a recent study revealed that treatment with sirolimus, an mTOR inhibitor, decreased MERS-CoV infection by more than 60% [103]. It has also been previously shown that optimal West Nile Virus (WNV) growth and protein expression are dependent on mTORC1-mediated activation of downstream signaling pathways, 4E-BP1 and eukaryotic initiation factor 4F (eIF4F) [125]. More importantly, a recent study aiming to identify drug combinations that may provide a synergistic effect in potentially treating SARS-CoV-2 with precise mechanism of action by network analysis revealed sirolimus plus dactinomycin as a potential drug combination for SARS-CoV-2 [126].
However, other studies have described an anti-viral role for mTOR. Recent findings have demonstrated that PI3K/AKT/mTOR signaling pathway is crucial for cytokine responses in IL-15 primed natural killer (NK) cells. Moreover, mTOR inhibition using rapamycin results in defects in both proliferation of NK cells and production of IFN-γ and granzyme B, leading to increased viral burdens upon murine cytomegalovirus infection [127]. These findings describe a link between the metabolic sensor mTOR and NK cell anti-viral responses. In addition, a recent study has shown that metformin, by upregulating the expression of AMPK and inhibiting mTOR-mediated pathway, decreases IFN-α expression following seasonal vaccination (SV) with trivalent influenza vaccine (TIV) and is associated with impaired antibody responses in patients with type 2 diabetes [128]. mTOR has also been described to play a role in suppressing hepatitis C virus (HCV) RNA replication, proposing that the activation of mTOR by HCV is an anti-viral response by the cells [129]. Taken together, these findings suggest that extensive research on the exact role of mTOR in viral infections is still strongly warranted.
On another note, the metabolic sensor mTOR is negatively regulated by Regulated in Development and DNA Damage Responses 1 (REDD1) [33]. IL-6, which was closely related to the occurrence of severe COVID-19 in adult patients, was shown to reduce basal as well as stress-induced REDD1 in a Signal Transducer and Activator of Transcription 3 (STAT3) dependent manner, resulting in the activation of mTOR [33]. Remarkably, rapamycin was shown to ameliorate IL-6-induced insulin resistance in liver cells [130]. Based on these observations, we suggest that COVID-19 associated cytokine storm might worsen the prognosis of DM by dysregulating the AMPK/mTOR signaling pathway (Fig. 3). Collectively, these observations suggest that activating AMPK and/or inhibiting mTOR-mediated signaling pathway could be used as novel drug targets for therapeutic intervention strategies.

Proposed crosstalk between COVID-19 and Diabetes Mellitus
5 Conclusion
In this review, we describe three potential mechanisms underlying the increased susceptibility of patients with diabetes to a more severe COVID-19 disease, leading to higher morbidity and mortality. Several studies have tried to explain a possible increased susceptibility to infection with SARS-CoV-2 in patients with diabetes. However, no data has shown, to date, that these patients are at higher risk of contracting COVID-19. SARS-CoV-2 enters the host cell through the ACE2 receptor. While a consensus has still not been reached on the role of ACE2 in the crosstalk between diabetes and COVID-19, some argue that patients with diabetes have an elevated ACE2 expression, thus facilitating viral entry and subsequent replication. Others show that patients with diabetes have low levels of ACE2 and that the observed increase in ACE2 is due to other factors such as treatment with ACE/ARBs, hypoglycemic agents and statins. Patients with diabetes present with elevated basal levels of cytokines, such as IL1-β and IL-6, and with a state of low-grade chronic inflammation that seems to further intensify the hyperinflammation observed in response to SARS-CoV-2. This so-called “cytokine storm”, particularly with the increase in IL-6, is also suggested to alter AMPK/mTOR signaling pathway in patients with diabetes, possibly aggravating insulin resistance and diabetes-induced complications. Therefore, despite the need for further research investigation, one can speculate that treatment with human recombinant soluble ACE2, IL-6 antagonists, AMPK activators or mTOR inhibitors may be considered as potential therapeutic strategies to alleviate and even halt the complications associated with COVID-19 disease.
References
de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–34. https://doi.org/10.1038/nrmicro.2016.81.
Organization WH. Middle East respiratory syndrome coronavirus (MERS-CoV)[WHO website]. 2019. 2020.
Nsoesie EO, Rader B, Barnoon YL, Goodwin L, Brownstein J. Analysis of hospital traffic and search engine data in Wuhan China indicates early disease activity in the fall of 2019. 2020.
Velavan TP, Meyer CG. The COVID-19 epidemic. Tropical Med Int Health. 2020;25(3):278–80.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239. https://doi.org/10.1001/jama.2020.2648.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–20. https://doi.org/10.1056/NEJMoa2002032.
Kass DA, Duggal P, Cingolani O. Obesity could shift severe COVID-19 disease to younger ages. Lancet. 2020;395(10236):1544–5. https://doi.org/10.1016/S0140-6736(20)31024-2.
Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–81. https://doi.org/10.1016/S2213-2600(20)30079-5.
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan. China JAMA Intern Med. 2020;180:934. https://doi.org/10.1001/jamainternmed.2020.0994.
Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–23.
Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan. China Intensive Care Medicine. 2020;46:846–8. https://doi.org/10.1007/s00134-020-05991-x.
Carlos WG, Dela Cruz CS, Cao B, Pasnick S, Jamil S. Novel Wuhan (2019-nCoV) coronavirus. Am J Respir Crit Care Med. 2020;201(4):P7–8. https://doi.org/10.1164/rccm.2014P7.
Ren LL, Wang YM, Wu ZQ, Xiang ZC, Guo L, Xu T, et al. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin Med J. 2020;133:1015–24. https://doi.org/10.1097/CM9.0000000000000722.
Wang W, Tang J, Wei F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan. China J Med Virol. 2020;92(4):441–7. https://doi.org/10.1002/jmv.25689.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Chung M, Bernheim A, Mei X, Zhang N, Huang M, Zeng X, et al. CT imaging features of 2019 novel coronavirus (2019-nCoV). Radiology. 2020;200230.
Ye Z, Zhang Y, Wang Y, Huang Z, Song B. Chest CT manifestations of new coronavirus disease 2019 (COVID-19): a pictorial review. Eur Radiol 2020:1–9.
Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7(6):439–50. https://doi.org/10.1038/nrmicro2147.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94(7). doi:https://doi.org/10.1128/JVI.00127-20.
Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237–61. https://doi.org/10.1146/annurev-virology-110615-042301.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–4. https://doi.org/10.1038/nature02145.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunologic features in severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620–9. https://doi.org/10.1172/JCI137244.
Yang M. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. Available at SSRN 3527420. 2020.
Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020;34(2). doi: https://doi.org/10.23812/CONTI-E.
Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, et al. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L341–53. https://doi.org/10.1152/ajplung.00122.2010.
Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. 2020;146:119–127.e4. https://doi.org/10.1016/j.jaci.2020.04.027.
Pugin J, Ricou B, Steinberg KP, Suter PM, Martin TR. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med. 1996;153(6 Pt 1):1850–6. https://doi.org/10.1164/ajrccm.153.6.8665045.
Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY, et al. Blockade of IL-6 trans signaling attenuates pulmonary fibrosis. J Immunol. 2014;193(7):3755–68. https://doi.org/10.4049/jimmunol.1302470.
Gong J, Dong H, Xia SQ, Huang YZ, Wang D, Zhao Y et al. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia. medRxiv. 2020:2020.02.25.20025643. doi:https://doi.org/10.1101/2020.02.25.20025643.
Campochiaro C, Della-Torre E, Cavalli G, De Luca G, Ripa M, Boffini N, et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: a single-Centre retrospective cohort study. Eur J Intern Med. 2020;76:43–9. https://doi.org/10.1016/j.ejim.2020.05.021.
Registry CCT. A multicenter, randomized controlled trial for the efficacy and safety of tocilizumab in the treatment of new coronavirus pneumonia (COVID-19). 2020.
l Cg. Tocilizumab in COVID-19 Pneumonia (TOCIVID-19)(TOCIVID-19). 2020.
Pinno J, Bongartz H, Klepsch O, Wundrack N, Poli V, Schaper F, et al. Interleukin-6 influences stress-signalling by reducing the expression of the mTOR-inhibitor REDD1 in a STAT3-dependent manner. Cell Signal. 2016;28(8):907–16. https://doi.org/10.1016/j.cellsig.2016.04.004.
Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136(1):95–103. https://doi.org/10.1111/j.1365-2249.2004.02415.x.
Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. 2006;6(3):271–6. https://doi.org/10.1016/j.coph.2006.03.001.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–6. https://doi.org/10.1038/nature03712.
Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93(5):543–8. https://doi.org/10.1113/expphysiol.2007.040048.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–9. https://doi.org/10.1038/nm1267.
Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84(2):1198–205. https://doi.org/10.1128/JVI.01248-09.
Wang S, Guo F, Liu K, Wang H, Rao S, Yang P, et al. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008;136(1–2):8–15. https://doi.org/10.1016/j.virusres.2008.03.004.
Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L84–96. https://doi.org/10.1152/ajplung.00071.2009.
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105(22):7809–14. https://doi.org/10.1073/pnas.0711241105.
Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. 2005;280(34):30113–9. https://doi.org/10.1074/jbc.M505111200.
Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. 2020. https://doi.org/10.1001/jama.2020.4683.
Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan. China Allergy. 2020;75:1730–41. https://doi.org/10.1111/all.14238.
Shah BR, Hux JE. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care. 2003;26(2):510–3. https://doi.org/10.2337/diacare.26.2.510.
Muller LM, Gorter KJ, Hak E, Goudzwaard WL, Schellevis FG, Hoepelman AI, et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. 2005;41(3):281–8. https://doi.org/10.1086/431587.
Joshi N, Caputo GM, Weitekamp MR, Karchmer AW. Infections in patients with diabetes mellitus. N Engl J Med. 1999;341(25):1906–12. https://doi.org/10.1056/NEJM199912163412507.
Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology. 2015;144(2):171–85. https://doi.org/10.1111/imm.12394.
Wu H, Lau ESH, Ma RCW, Kong APS, Wild SH, Goggins W, et al. Secular trends in all-cause and cause-specific mortality rates in people with diabetes in Hong Kong, 2001-2016: a retrospective cohort study. Diabetologia. 2020;63(4):757–66. https://doi.org/10.1007/s00125-019-05074-7.
Yang JK, Feng Y, Yuan MY, Yuan SY, Fu HJ, Wu BY, et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet Med. 2006;23(6):623–8. https://doi.org/10.1111/j.1464-5491.2006.01861.x.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239–42. https://doi.org/10.1001/jama.2020.2648.
Emami A, Javanmardi F, Pirbonyeh N, Akbari A. Prevalence of underlying diseases in hospitalized patients with COVID-19: a systematic review and meta-analysis. Arch Acad Emerg Med. 2020;8(1):e35.
Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev. 2020:e3319. doi:https://doi.org/10.1002/dmrr.3319.
Stokes EK, Zambrano LD, Anderson KN, Marder EP, Raz KM, El Burai FS, et al. Coronavirus Disease 2019 Case Surveillance - United States, January 22–May 30, 2020. MMWR Morb Mortal Wkly Rep. 2020;69(24):759–65. https://doi.org/10.15585/mmwr.mm6924e2.
Fadini GP, Morieri ML, Longato E, Avogaro A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J Endocrinol Investig. 2020;43(6):867–9. https://doi.org/10.1007/s40618-020-01236-2.
Muniyappa R, Gubbi S. COVID-19 pandemic, Corona viruses, and diabetes mellitus. Am J Physiol Endocrinol Metab. 2020;318:E736–41. https://doi.org/10.1152/ajpendo.00124.2020.
Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. 1999;26(3–4):259–65. https://doi.org/10.1111/j.1574-695X.1999.tb01397.x.
Jafar N, Edriss H, Nugent K. The effect of short-term hyperglycemia on the innate immune system. Am J Med Sci. 2016;351(2):201–11. https://doi.org/10.1016/j.amjms.2015.11.011.
Ma RCW, Holt RIG. COVID-19 and diabetes. Diabet Med. 2020;37:723–5. https://doi.org/10.1111/dme.14300.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605–10. https://doi.org/10.1161/CIRCULATIONAHA.104.510461.
Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43(5):970–6. https://doi.org/10.1161/01.HYP.0000124667.34652.1a.
Liu F, Long X, Zhang B, Zhang W, Chen X, Zhang Z. ACE2 expression in pancreas May cause pancreatic damage after SARS-CoV-2 infection. Clin Gastroenterol Hepatol. 2020;18:2128–2130.e2. https://doi.org/10.1016/j.cgh.2020.04.040.
Lukassen S, Lorenz Chua R, Trefzer T, Kahn NC, Schneider MA, Muley T et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020. doi:https://doi.org/10.15252/embj.20105114.
Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14:185–92. https://doi.org/10.1007/s11684-020-0754-0.
Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, et al. ACE and ACE2 activity in diabetic mice. Diabetes. 2006;55(7):2132–9. https://doi.org/10.2337/db06-0033.
Roca-Ho H, Riera M, Palau V, Pascual J, Soler MJ. Characterization of ACE and ACE2 Expression within Different Organs of the NOD Mouse. Int J Mol Sci. 2017;18(3). doi:https://doi.org/10.3390/ijms18030563.
Rao S, Lau A, So HC. Exploring diseases/traits and blood proteins causally related to expression of ACE2, the putative receptor of SARS-CoV-2: a Mendelian randomization analysis highlights tentative relevance of diabetes-related traits. Diabetes Care. 2020;43(7):1416–26. https://doi.org/10.2337/dc20-0643
Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008;74(12):1610–6. https://doi.org/10.1038/ki.2008.497.
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020. https://doi.org/10.1016/j.cell.2020.04.004.
Ohishi M. Hypertension with diabetes mellitus: physiology and pathology. Hypertens Res. 2018;41(6):389–93. https://doi.org/10.1038/s41440-018-0034-4.
Fountain JH, Lappin SL. Physiology. Treasure Island (FL): Renin Angiotensin System. StatPearls; 2020.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21. https://doi.org/10.1016/S2213-2600(20)30116-8.
Romani-Perez M, Outeirino-Iglesias V, Moya CM, Santisteban P, Gonzalez-Matias LC, Vigo E, et al. Activation of the GLP-1 receptor by Liraglutide increases ACE2 expression, reversing right ventricle hypertrophy, and improving the production of SP-A and SP-B in the lungs of type 1 diabetes rats. Endocrinology. 2015;156(10):3559–69. https://doi.org/10.1210/en.2014-1685.
Tikoo K, Patel G, Kumar S, Karpe PA, Sanghavi M, Malek V, et al. Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: role of epigenetic histone modifications. Biochem Pharmacol. 2015;93(3):343–51. https://doi.org/10.1016/j.bcp.2014.11.013.
Zhang W, Xu YZ, Liu B, Wu R, Yang YY, Xiao XQ, et al. Pioglitazone upregulates angiotensin converting enzyme 2 expression in insulin-sensitive tissues in rats with high-fat diet-induced nonalcoholic steatohepatitis. Scientific World J. 2014;2014:603409–7. https://doi.org/10.1155/2014/603409.
Organization WH. The use of non-steroidal anti-inflammatory drugs (NSAIDs) in patients with COVID-19: scientific brief, 19 April 2020: World Health Organization2020.
Fernandez C, Rysa J, Almgren P, Nilsson J, Engstrom G, Orho-Melander M, et al. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J Intern Med. 2018;284(4):377–87. https://doi.org/10.1111/joim.12783.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78:779–784.e5.
Pal R, Bhansali A. COVID-19, diabetes mellitus and ACE2: the conundrum. Diabetes Res Clin Pract. 2020;162:108132. https://doi.org/10.1016/j.diabres.2020.108132.
Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res. 2020. https://doi.org/10.1002/ddr.21656.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90. https://doi.org/10.1007/s00134-020-05985-9.
Batlle D, Wysocki J, Satchell K. Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci. 2020;134(5):543–5. https://doi.org/10.1042/CS20200163.
Chee YJ, Ng SJH, Yeoh E. Diabetic ketoacidosis precipitated by Covid-19 in a patient with newly diagnosed diabetes mellitus. Diabetes Res Clin Pract. 2020;164:108166. https://doi.org/10.1016/j.diabres.2020.108166.
Li J, Wang X, Chen J, Zuo X, Zhang H, Deng A. COVID-19 infection may cause ketosis and ketoacidosis. Diabetes Obes Metab. 2020. https://doi.org/10.1111/dom.14057.
Ren H, Yang Y, Wang F, Yan Y, Shi X, Dong K, et al. Association of the insulin resistance marker TyG index with the severity and mortality of COVID-19. Cardiovasc Diabetol. 2020;19(1):58. https://doi.org/10.1186/s12933-020-01035-2.
Rubino F, Amiel SA, Zimmet P, Alberti G, Bornstein S, Eckel RH, et al. New-onset diabetes in Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMc2018688.
Bindom SM, Lazartigues E. The sweeter side of ACE2: physiological evidence for a role in diabetes. Mol Cell Endocrinol. 2009;302(2):193–202. https://doi.org/10.1016/j.mce.2008.09.020.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7. https://doi.org/10.1002/path.1570.
Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European prospective investigation into Cancer and nutrition (EPIC)-Potsdam study. Diabetes. 2003;52(3):812–7. https://doi.org/10.2337/diabetes.52.3.812.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–4. https://doi.org/10.1016/S0140-6736(20)30628-0.
Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. https://doi.org/10.1038/nrc3245.
Imani F, Horii Y, Suthanthiran M, Skolnik EY, Makita Z, Sharma V, et al. Advanced glycosylation endproduct-specific receptors on human and rat T-lymphocytes mediate synthesis of interferon gamma: role in tissue remodeling. J Exp Med. 1993;178(6):2165–72. https://doi.org/10.1084/jem.178.6.2165.
Morohoshi M, Fujisawa K, Uchimura I, Numano F. The effect of glucose and advanced glycosylation end products on IL-6 production by human monocytes. Ann N Y Acad Sci. 1995;748:562–70. https://doi.org/10.1111/j.1749-6632.1994.tb17362.x.
Vlassara H, Brownlee M, Manogue KR, Dinarello CA, Pasagian A. Cachectin/TNF and IL-1 induced by glucose-modified proteins: role in normal tissue remodeling. Science. 1988;240(4858):1546–8. https://doi.org/10.1126/science.3259727.
Kuwabara WMT, Yokota CNF, Curi R, Alba-Loureiro TC. Obesity and type 2 diabetes mellitus induce lipopolysaccharide tolerance in rat neutrophils. Sci Rep. 2018;8(1):17534. https://doi.org/10.1038/s41598-018-35809-2.
Peleg AY, Weerarathna T, McCarthy JS, Davis TM. Common infections in diabetes: pathogenesis, management and relationship to glycaemic control. Diabetes Metab Res Rev. 2007;23(1):3–13. https://doi.org/10.1002/dmrr.682.
Ziegler-Heitbrock HW, Wedel A, Schraut W, Strobel M, Wendelgass P, Sternsdorf T, et al. Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J Biol Chem. 1994;269(25):17001–4.
Geerlings SE, Brouwer EC, Van Kessel KC, Gaastra W, Stolk RP, Hoepelman AI. Cytokine secretion is impaired in women with diabetes mellitus. Eur J Clin Investig. 2000;30(11):995–1001. https://doi.org/10.1046/j.1365-2362.2000.00745.x.
Kulcsar KA, Coleman CM, Beck SE, Frieman MB. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight. 2019;4(20). doi:https://doi.org/10.1172/jci.insight.131774.
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–2. https://doi.org/10.1016/S2213-2600(20)30076-X.
Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents. 2020;105954:105954. https://doi.org/10.1016/j.ijantimicag.2020.105954.
Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015;59(2):1088–99. https://doi.org/10.1128/AAC.03659-14.
Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. https://doi.org/10.1146/annurev-pharmtox-010611-134537.
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–68. https://doi.org/10.1016/s1097-2765(02)00636-6.
Choi KM, McMahon LP, Lawrence JC Jr. Two motifs in the translational repressor PHAS-I required for efficient phosphorylation by mammalian target of rapamycin and for recognition by raptor. J Biol Chem. 2003;278(22):19667–73. https://doi.org/10.1074/jbc.M301142200.
Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S. Yoshino K-i et al. the mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–4.
Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13(10):797–806. https://doi.org/10.1016/s0960-9822(03)00329-4.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. https://doi.org/10.1126/science.1106148.
Eid AA, Ford BM, Bhandary B, de Cassia CR, Block K, Barnes JL, et al. Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes. 2013;62(8):2935–47. https://doi.org/10.2337/db12-1504.
Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol. 2013;33(17):3439–60. https://doi.org/10.1128/MCB.00217-13.
Eid S, Boutary S, Braych K, Sabra R, Massaad C, Hamdy A, et al. mTORC2 signaling regulates Nox4-induced Podocyte depletion in diabetes. Antioxid Redox Signal. 2016;25(13):703–19. https://doi.org/10.1089/ars.2015.6562.
Eid S, Maalouf R, Jaffa AA, Nassif J, Hamdy A, Rashid A, et al. 20-HETE and EETs in diabetic nephropathy: a novel mechanistic pathway. PLoS One. 2013;8(8):e70029. https://doi.org/10.1371/journal.pone.0070029.
Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764–72. https://doi.org/10.1172/JCI67227.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. https://doi.org/10.1172/JCI13505.
Livingstone R, Boyle JG, Petrie JR, Team RS. A new perspective on metformin therapy in type 1 diabetes. Diabetologia. 2017;60(9):1594–600. https://doi.org/10.1007/s00125-017-4364-6.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. https://doi.org/10.1016/j.molcel.2008.03.003.
Pepin E, Al-Mass A, Attane C, Zhang K, Lamontagne J, Lussier R, et al. Pancreatic beta-cell dysfunction in diet-induced obese mice: roles of AMP-kinase, protein kinase Cepsilon, mitochondrial and cholesterol metabolism, and alterations in gene expression. PLoS One. 2016;11(4):e0153017. https://doi.org/10.1371/journal.pone.0153017.
Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3(4):340–51. https://doi.org/10.1038/nrd1344.
Sun Y, Ren M, Gao GQ, Gong B, Xin W, Guo H, et al. Chronic palmitate exposure inhibits AMPKalpha and decreases glucose-stimulated insulin secretion from beta-cells: modulation by fenofibrate. Acta Pharmacol Sin. 2008;29(4):443–50. https://doi.org/10.1111/j.1745-7254.2008.00717.x.
Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D. Metformin improves in vivo and in vitro B cell function in individuals with obesity and Type-2 diabetes. Vaccine. 2017;35(20):2694–700. https://doi.org/10.1016/j.vaccine.2017.03.078.
Stohr S, Costa R, Sandmann L, Westhaus S, Pfaender S. Anggakusuma et al. host cell mTORC1 is required for HCV RNA replication. Gut. 2016;65(12):2017–28. https://doi.org/10.1136/gutjnl-2014-308971.
McNulty S, Flint M, Nichol ST, Spiropoulou CF. Host mTORC1 signaling regulates Andes virus replication. J Virol. 2013;87(2):912–22. https://doi.org/10.1128/JVI.02415-12.
Wang CH, Chung FT, Lin SM, Huang SY, Chou CL, Lee KY, et al. Adjuvant treatment with a mammalian target of rapamycin inhibitor, sirolimus, and steroids improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure. Crit Care Med. 2014;42(2):313–21. https://doi.org/10.1097/CCM.0b013e3182a2727d.
Shives KD, Massey AR, May NA, Morrison TE, Beckham JD. 4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression. Viruses. 2016;8(10). doi:https://doi.org/10.3390/v8100287.
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. https://doi.org/10.1038/s41421-020-0153-3.
Nandagopal N, Ali AK, Komal AK, Lee SH. The critical role of IL-15-PI3K-mTOR pathway in natural killer cell effector functions. Front Immunol. 2014;5:187. https://doi.org/10.3389/fimmu.2014.00187.
Saenwongsa W, Nithichanon A, Chittaganpitch M, Buayai K, Kewcharoenwong C, Thumrongwilainet B, et al. Metformin-induced suppression of IFN-alpha via mTORC1 signalling following seasonal vaccination is associated with impaired antibody responses in type 2 diabetes. Sci Rep. 2020;10(1):3229. https://doi.org/10.1038/s41598-020-60213-0.
Johri MK, Lashkari HV, Gupta D, Vedagiri D, Harshan KH. mTORC1 restricts hepatitis C virus RNA replication through ULK1-mediated suppression of miR-122 and facilitates post-replication events. J Gen Virol. 2020;101(1):86–95. https://doi.org/10.1099/jgv.0.001356.
Kim JH, Kim JE, Liu HY, Cao W, Chen J. Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem. 2008;283(2):708–15. https://doi.org/10.1074/jbc.M708568200.
Acknowledgments
We would like to thank the President of the American University of Beirut, Administration, Faculty Members and Staff of this great institution for their support in this time of crisis.
Funding
RN is funded by the American University of Beirut Faculty of Medicine Biomedical Sciences graduate program. STA and AAE are supported by the American University of Beirut Medical Practice Plan regular research grant. This content is solely the responsibility of the authors. The funding sources had no involvement in any aspect of the research.
Figures created with BioRender.com
Author information
Authors and Affiliations
Contributions
AAE conceived and designed the review article and approved the final version to be submitted. WSA, RN, AHF and NSA performed the literature review; analyzed and interpreted the data; wrote multiple subsections of the manuscript; and revised the manuscript for intellectual content. STA and MES reviewed and improved the entire manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Azar, W.S., Njeim, R., Fares, A.H. et al. COVID-19 and diabetes mellitus: how one pandemic worsens the other. Rev Endocr Metab Disord 21, 451–463 (2020). https://doi.org/10.1007/s11154-020-09573-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-020-09573-6