
Abstract
Somatostatin (SS) and dopamine (DA) receptors are widely expressed in neuroendocrine tumours that cause Cushing’s Syndrome (CS). Increasing knowledge of specific subtype expression within these tumours and the ability to target these receptor subtypes with high-affinity compounds, has driven the search for new SS- or DA-based medical therapies for the various forms of CS. In Cushing’s disease, corticotroph adenomas mainly express dopamine receptor subtype 2 (D2) and somatostatin receptor subtype 5 (sst5), whereas sst2 is expressed at lower levels. Activation of these receptors can inhibit ACTH-release in primary cultured corticotroph adenomas and compounds that target either sst5 (pasireotide, or SOM230) or D2 (cabergoline) have shown significant efficacy in subsets of patients in recent clinical studies. Combination therapy, either by administration of both types of compounds separately or by treatment with novel somatostatin–dopamine chimeric molecules (e.g. BIM-23A760), appears to be a promising approach in this respect. In selected cases of Ectopic ACTH-producing Syndrome (EAS), the sst2-preferring compound octreotide is able to reduce cortisol levels effectively. A recent study showed that D2 receptors are also significantly expressed in the majority of EAS and that cabergoline may decrease cortisol levels in subsets of these patients. In both normal adrenal tissue as well as in adrenal adenomas and carcinomas that cause CS, sst and DA receptor expression has been demonstrated. Although selected cases of adrenal CS may benefit from sst or DA-targeted treatment, its total contribution to the treatment of these patients is likely to be low as surgery is effective in most cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cushing’s syndrome (CS) is a severe endocrine condition, in which chronic systemic hypercortisolism leads to a variety of signs and symptoms such as centripetal obesity, peripheral muscle wasting, hypertension, diabetes mellitus, osteoporosis and psychiatric disturbances [1]. In untreated cases, morbidity and mortality rates are significantly elevated compared to those in normal subjects [2]. Around 70% of endogenous cases of CS are due to a pituitary ACTH-producing basophilic adenoma (Cushing’s disease, CD), around 15% are due to an adrenal cortisol-producing benign or malignant tumour and the remaining cases are generally due to ectopic ACTH-secretion from a neuro-endocrine tumour elsewhere in the body [3]. First-line treatment of Cushing’s syndrome, regardless of its origin, is usually surgery with the attempt to radically remove the ACTH- or cortisol-producing tumour.
However, not all cases of CS are well managed by surgery alone. For patients with CD it is known that long-term remission rates after transsphenoidal adenomectomy decrease to 50–80% with longer follow-up time [4]. A second transsphenoidal surgery in patients with a recurrence of CD is known to have significantly lower success rates than the primary surgery [5, 6]. Other treatment options for patients with persistent or recurrent CD include conventional radiotherapy or gamma knife surgery. Both are effective at reducing ACTH hypersecretion in the majority of patients, but have a slow onset of action, with an average time until remission of 9–24 months [7]. In addition, radiotherapy is accompanied by a significant risk of inducing secondary pituitary dysfunction, cranial nerve damage or secondary brain tumours [7–9]. As a definitive cure, patients with CD can undergo bilateral adrenalectomy, but this does have important implications in terms of lifelong dependence on adrenal hormone replacement therapy and risk of future Addison crises, while there is risk of the development of Nelson’s syndrome.
Similarly, in some patients with the ectopic ACTH-producing syndrome (EAS), the primary neuro-endocrine tumour cannot be localized despite extensive radiological and nuclear imaging, whereas in other EAS patients, tumour resection can be irradical [10, 11]. Both types of EAS patients will benefit from bilateral adrenalectomy but with the same disadvantages as described above for CD. Also, some patients with CS due to a malignant adrenal tumour have advanced disease at the time of presentation and in these cases complete removal of the cortisol-producing adrenal tumour will not be possible.
For the above reasons there is a clear rationale behind the search for an effective medical therapy for all causes of CS. A great number of drugs that act at the level of the pituitary, the adrenals or the glucocorticoid receptor itself, have been evaluated in the past decades with generally modest and variable results [12]. Most compounds either show limited efficacy or are associated with serious toxicity and adverse events. For instance, the steroidogenic inhibitor ketoconazole has been shown to effectively decrease cortisol levels in about 50% of patients [13], but often causes considerable gastro-intestinal side effects and carries a serious risk of medication-induced hepatitis, which limits its use as a long-term monotherapy in CD patients [14]. Similarly, metyrapone can be effective in reducing cortisol levels in patients with CS, but can cause hypertension and hypokalemia. Blockade of the glucocorticoid receptor with mifepristone (RU-486) can improve symptoms of CS, but the absence of a suitable biochemical parameter to monitor treatment efficacy makes dose titration difficult and this can result in severe adrenal insufficiency in some patients.
In recent years, however, research into medical therapies for CS has also focussed on the use of both somatostatin (SS) and dopamine (DA) agonists. These are regarded as neuromodulatory agents that can inhibit ACTH-hypersecretion at the level of the pituitary. Important new insights in SS and DA receptor physiology, combined with the recent availability of more selective SS and DA-agonists, have emerged and are the topic of the current review.
2 Somatostatin and dopamine receptors
2.1 Somatostatin and dopamine receptors
Somatostatin (SS) is a 14 amino acid-long cyclic peptide that is widely distributed throughout the human body. Its functions vary from increasing gastro-intestinal motility to neurotransmission within the central nervous system, mediating immune responses and inhibition of hormone release [15, 16]. SS exerts its functions by binding to all five somatostatin receptor subtypes (sst1–5), which belong to the family of G-protein coupled receptors (GPCRs) [17]. Dopamine (DA) is a catecholamine with an equally wide range of functions including neurotransmission, control of vascular tone, renal function and hormone secretion [18]. Also for DA receptors, five subtypes are known (D1–5) that belong to the GPCR family, which are further classified into D1-like (D1, D5) and D2-like (D2, D3, D4). D1-like receptors generally mediate stimulatory functions, whereas most D2-like receptors are associated with inhibition. Upon binding of SS or DA to their respective receptors expressed on the plasma membrane of target cells, multiple cellular effector systems can be activated, which include inhibition of Ca2+-influx, inhibition of adenylyl cyclase activity or stimulation of phosphotyrosine phosphatases [17, 18]. Both sst and DA receptors are abundantly expressed in the human neuro-endocrine system and in the tumours that are derived from it [19, 20]. Most of the in vivo functions of SS and DA (D2-like) receptors are inhibitory and, therefore, targeting these receptors with their natural agonists or synthetically derived analogs has provided opportunities for medical therapy of various neuro-endocrine disorders.
2.2 SS analogs and DA agonists
Soon after its discovery in 1972, somatostatin was known to be a major regulator of GH release from the pituitary and was therefore of interest for the potential treatment of acromegaly [21]. One of the characteristics of native SS, however, is its very short half-life in the circulation, which is approximately 3 min [22]. For that reason, the production of synthetic SS analogs with a significantly longer half-life, was a major step forward in the treatment of this disorder. The first stable SS-analog produced was Octreotide (SMS 201–995), which has a half life of approximately 120 min after subcutaneous administration and was shown to reduce GH and IGF-1 levels in approximately two thirds of acromegalic patients [23, 24].
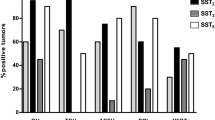
Another important step in the development of SS analogs was the discovery of the 5 somatostatin receptor subtypes in the 1990s. These findings clarified indirectly that the two available SS-analogs at the time, Octreotide and its long-acting form Lanreotide, bind preferentially to the sst2, but only modestly to sst5 or any of the other subtypes. Native SS, on the other hand, binds with high affinity to all of its receptors (sst1–5). In the subsequent years, evidence grew that not all neuro-endocrine tumours expressed receptor subtypes in a similar manner. Whereas growth-hormone producing adenomas generally expressed high levels of sst2, other adenomas, such as corticotroph adenomas expressed considerably lower levels of sst2 (see Fig. 1). The concept that different neuro-endocrine tumours with important differences in sst expression profiles would require specific sst-targeting analogs, sparked the interest for the development of new types of SS-analogs that displayed high affinity for one or more SS receptor subtypes, see Table 1. One example of such a compound is BIM-23244, which is a bispecific SS-analog with high affinity for both sst2 and sst5. In GH-producing adenomas that were only partially responsive to Octreotide, this novel bispecific compound suppressed GH-production in vitro significantly more effective than Octreotide, probably through co-activation of sst5 receptors [25]. Another example is Pasireotide (SOM230), which is a multi-ligand SS-analog with high binding affinity for the sst1, sst2, sst3 and sst5 with IC50 values of 9.3, 1.0, 1.5 and 0.16 nM, respectively, see Table 1 [26]. Its binding profile, which includes high sst5-affinity, makes it a promising new drug in the treatment of a number of neuro-endocrine tumours, including CD (see below).

Overview of sst expression in seven GH-producing (somatotroph) and six ACTH-producing (corticotroph) human pituitary adenomas. Note the difference in scale of the y-axis between sst1–3 and sst5. Somatotroph adenomas have abundant expression of both sst2 and sst5 (left column), whereas corticotroph adenomas express sst5 at similar levels but have a significantly lower expression of sst2 (right column). Values are expressed as copy numbers relative to that of the reference gene hprt. N.d. Not detectable (Adapted from [51, 115])
Dopamine agonists are an important class of drugs with a broad range of therapeutic indications, including neurological disorders (Parkinson’s disease), cardiovascular dysfunction and neuro-endocrine disorders. They can be classified into either non-ergot (e.g. quinagolide) or ergot-derived (e.g. bromocriptine, cabergoline, pergolide). Bromocriptine has been known for many years to effectively inhibit prolactin (PRL) release in the majority of prolactinomas [27]. With increasing knowledge on DA receptors, it also became evident that selectivity of dopaminergic compounds for DA receptor subtypes was of great importance for their overall efficacy and safety profile. In comparison with bromocriptine, cabergoline has a longer plasma half-life, binds with a higher affinity to the D2 receptor, is better tolerated by patients and can induce normalization in patients with hyperprolactinemia that are proven to be resistant to bromocriptine therapy [28]. The fulfilment of these criteria makes cabergoline a promising drug for the treatment of a number of neuro-endocrine disorders in which D2 expression plays an imminent role.
2.3 Chimeric somatostatin–dopamine compounds
The fact that many neuro-endocrine cells co-express both sst and DA receptors, has driven the hypothesis that these receptors may work synergistically. In 2000, Rocheville et al. [29] published an important paper on the functional heterodimerization of sst5 and D2 receptors in stably transfected CHO-K1 cells, which resulted in overall enhanced biological potency. Based on these observations, new chimeric molecules have been synthesized that contain structural elements of both SS and DA compounds and therefore bind with high affinity to both sst and DA receptor subtypes. By binding to the two different receptors, these hybrid molecules may draw the receptors together in a spatial manner. This can lead to enhanced potency of the chimeric compound, compared to activation by two separate DA or SS analogs [30].
3 Cushing’s disease (CD)
3.1 Somatostatin analogs in Cushing’s disease
3.1.1 Sst expression in normal corticotroph cells
Whereas the role of hypothalamic SS as a principal regulator of pituitary GH-release has been firmly established [31], the effect of SS on ACTH release by the anterior pituitary gland has been less clear. Rat pituitary corticotrophs are known to express multiple sst, including sst2 and sst5 [32–34], but treatment of cultured rat corticotrophs with SS-14 does not result in inhibition of ACTH-release [35, 36]. On the other hand, when rat pituitary cells are cultured in glucocorticoid-free media, SS-14 is able to decrease ACTH-release [37]. In agreement with these findings, infusion of SS-14 or Octreotide does not alter ACTH-release in normal subjects [38–41], whereas both of these compounds can acutely decrease plasma ACTH levels in conditions of hypocortisolemia such as untreated Addison disease [42]. These observations suggest that the presence of glucocorticoids reduces the inhibitory effects of native somatostatin and traditional SS analogs on ACTH release.
3.1.2 In vitro studies with SS-analogs in corticotroph cell lines and adenomas
The only available ACTH producing cell line from corticotroph origin is the murine AtT20 cell line. A number of studies have indicated that in these cells sst2 and sst5 are principally involved in regulation of ACTH release and that selective agonists that target these subtypes effectively inhibit ACTH secretion [43–47]. More recently it was found that especially sst5 played a crucial role in regulating ACTH release in these cells and that sst5-targeting agonists were more effective than sst2-agonists in inhibiting ACTH release [48]. Interestingly, pre-incubation with dexamethasone decreased the expression of sst2 in these cells, but not of sst5, and in line with these findings Octreotide, but not Pasireotide, lost most of its ACTH inhibiting potential after glucocorticoid pre-treatment [48]. These data are in line with the original observations that glucocorticoids downregulate the total number of SS binding sites in cultured pituitary cells [49]. Evidence for abrogation of sst2-mediated effects by glucocorticoids has also been provided by Stalla et al. [50]. They found that Octreotide decreased ACTH levels in corticotroph adenomas in vitro, but not in CD patients in vivo. However, when these corticotroph adenoma cells in vitro were pre-treated with the glucocorticoid hydrocortisone, the ACTH inhibiting effects of Octreotide were abolished in one of the cultures. Given the generalized state of hypercortisolism in CD patients and the relative resistance of sst5 to glucocorticoid-induced down-regulation compared to sst2, SS-analogs with high sst5 affinity are of great interest in the development of new medical therapies for CD.
In 2005 and 2006, two studies were published that independently investigated sst expression in human corticotroph adenoma tissues, obtained at the time of transsphenoidal surgery. In the first study, Hofland et al. [51] showed by quantitative PCR that sst5 was highly expressed in 6/6 adenomas, whereas sst1,2,3,4 were expressed at much lower levels. In concordance with this, functional studies in five additional adenomas demonstrated overall superior ACTH inhibition by Pasireotide (10 nM) compared to Octreotide (10 nM) after 72 h.
In the second study, Batista et al. reported on a series of 13 corticotroph adenomas derived from both adult (n = 7) and pediatric (n = 6) CD patients [52]. In this study, quantitative PCR demonstrated the expression of subtypes 1, 2, 4 and 5 in these adenomas, while at immunohistochemistry expression of all subtypes was found. Both of these methods showed the highest expression of the sst5 subtype. Six of the adenomas were cultured in vitro and treated with Pasireotide. In 6/6 adenomas Pasireotide significantly decreased cellular proliferation rates (range 10–70%) as measured by uptake of fluorescent vital stain and in 5/6 a significant decrease in ACTH production was observed (range 23–56%) at doses of 1 to 10 nM after 48–96 h. Furthermore, a dissociation was seen in some of the adenomas between the anti-secretory and anti-proliferative effects of Pasireotide, similarly to what has been described previously for GH-producing adenomas in response to SS-analog treatment [53].
3.1.3 Clinical studies with SS-analogs in CD
Early studies showed that in patients with CD, Octreotide is not able to effectively reduce ACTH secretion and hence cortisol levels [50, 54, 55]. In contrast, several smaller studies and case reports found that patients with Nelson syndrome, i.e. an expanding ACTH-producing pituitary adenoma after bilateral adrenalectomy, did respond to Octreotide with reductions in ACTH [54, 56–58]. This difference is readily explained by the differences in average circulating cortisol levels in both disease states and the effects of glucocorticoid-induced down-regulation of sst2 receptors, as mentioned earlier [59].
Since then, few clinical studies have been reported that examined SS-analog therapy in CD, until some important new insights developed. It was foremost the discovery that sst5 was highly expressed in the majority of human corticotroph adenomas, which made the novel multi-ligand SS-analog Pasireotide an interesting compound to evaluate in patients with CD, due to its subnanomolar sst5-affinity. A phase II multi-center clinical study was performed in 21 patients with de novo or recurrent CD [60]. Patients were treated with SOM230 600 μg twice daily over a 15-day period. Primary endpoint was normalization of 24-h urinary free cortisol (UFC) levels. Preliminary results of this study showed that out of 21 included patients, four (19%) obtained complete UFC normalization and another five (24%) had a significant decrease in UFC. Overall, Pasireotide was well tolerated in the 2 × 600 μg dose, except for some mild gastro-intestinal side effects such as nausea, abdominal pain and loose stools or diarrhoea. A major side effect of Pasireotide, however, which was already known from previous studies in acromegalic patients, was an overall increase in blood glucose levels. Worsening of glycaemic control was observed in almost all participating patients in this study, but was often a transient effect. In some cases, however, the use of subcutaneous insulin administration was required and in one case the study drug was withdrawn.
Based on the current data, Pasireotide appears to be a potent cortisol-lowering drug in almost half of CD patients and is well tolerated. A major advantage of Pasireotide compared to Octreotide is its longer plasma half-life in the circulation, approximately 12 versus 2 h [61]. Pasireotide treatment does carry the risk of significant hyperglycemia, however, in a substantial number of patients. A new challenge would be to identify subgroups of CD patients who are likely to benefit from sst5-mediated (Pasireotide) therapy and at the same time do not suffer excessively from increases in blood glucose levels.
Another potential problem might be any direct effects of Pasireotide therapy on GH and IGF-1evels in CD patients. It is well known that Pasireotide, via sst5 receptor activation, directly decreases GH release and hence IGF-1 levels in patients with acromegaly. Whether this also occurs in normal subjects without GH/IGF-1 axis overactivation, has not been reported. Normal somatotrophs, however, express sst5 in significant numbers and in normal rats, primates and dogs Pasireotide has been shown to significantly decrease GH and IGF-1 levels [62]. In patients with CD, sustained hypercortisolism by itself causes a state of relative growth-hormone deficiency and therefore these patients may be at greater risk to become GH-deficient. Current and future clinical studies with Pasireotide in CD patients should therefore include careful investigation of the effects on the GH/IGF-1 axis.
3.2 Dopamine agonists in Cushing’s disease
3.2.1 DA receptor expression in normal corticotrophs
In humans, no firm data exist whether or not ACTH release is directly regulated by DA receptors in normal corticotroph cells. In rats it is known that the intermediate lobe in the pituitary is under tonic inhibitory control from dopaminergic neurons from the hypothalamus [63–65]. The predominant cell type in the intermediate lobe is the melanotroph, which produces pro-opiomelanocortin (POMC). In the intermediate lobe POMC is processed into α-melanocyte stimulating hormone (α-MSH) and corticotropin-like intermediate lobe peptide (CLIP). This is different from the POMC-processing in anterior corticotroph cells, which mainly results in ACTH. The tonic inhibition by hypothalamic dopamine is thought to be exerted through D2 receptors. This is demonstrated by the fact that D2-deficient mice develop intermediate lobe hypertrophy with increased POMC expression, elevated ACTH and corticosterone levels, resulting in adrenal gland hypertrophy [66]. In humans, the intermediate lobe in the pituitary is a rudimentary structure, but is still thought to contain important biological functions. Human corticotroph adenomas arising from the intermediate lobe may have different characteristics than those arising from the anterior lobe, although much controversy exists around this subject [67, 68].
3.2.2 In vitro studies with DA agonists in corticotroph cell lines and adenomas
Two reports have been published on the use of dopamine agonists in the murine corticotroph cell line AtT20, but these have produced conflicting results. Farrell et al. found that treatment with the dopamine agonist bromocriptine did not reduce POMC mRNA expression in these cells, whereas Yin et al. did show that bromocriptine inhibited proliferation of these cells with induction of apoptosis [69, 70]. The observed difference may be due to the fact that in the second study treatment with bromocriptine was significantly longer than in the first study (72 vs. 24 h, respectively).
In 2004, Pivonello et al. [71] investigated DA receptor expression in a series of 20 human corticotroph adenomas. They showed that the majority (80%) of these adenomas express the D2 receptor as demonstrated by immunohistochemistry (IHC), receptor-ligand binding and RT-PCR. Of these D2-positive adenomas, approximately 40% expressed the D2 long isoform, 20% D2 short and 40% expressed both isoforms. D4 was expressed in 20% of cases, whereas D1, D3 and D5 expression was not observed. Functional studies in vitro correlated very well with the D2 expression data: adenomas high in D2 expression responded well to either bromocriptine or cabergoline therapy with inhibition of ACTH release by 43% to 60%, whereas D2-negative adenomas failed to respond. The D2- expression data reported in this study are similar to those described by an earlier paper, where 11/16 (69%) of corticotroph adenomas, both functional and silent, expressed D2 receptors as demonstrated by in situ hybridisation and immunohistochemistry [72].
3.2.3 Clinical studies with DA agonists in CD
The DA agonist bromocriptine has been widely evaluated for its potential use in human corticotroph adenomas. Overall, results of these studies have been variable. Although initial reductions in ACTH levels are evident in almost half of CD patients in response to bromocriptine administration, these reductions are often minor and sustained responses to bromocriptine therapy occur only in a small percentage of patients [73]. Some studies have suggested that corticotroph adenomas arising from the intermediate lobe may be more likely to respond to bromocriptine [74].
Compared to bromocriptine, cabergoline binds with even higher specificity and affinity to D2-receptors and has a longer duration of action [28]. Over the past decade, various case reports have demonstrated that ACTH-producing adenomas can be highly responsive to cabergoline therapy, both in patients with CD as well as in Nelson’s syndrome [71, 75–83], see Table 2. In some of these cases shrinkage of the corticotroph adenoma was observed on MRI [75–78, 82]. In the previously mentioned study by Pivonello et al., 20 patients with CD were treated with cabergoline at a dose of 1–3 mg/week for 3 months [71]. This resulted in a significant decrease in urinary free cortisol (UFC) in 60% of patients and even complete UFC normalization in 40% of them. Interestingly, there was a very good correlation between the in vitro findings on D2 receptor expression and the responses to cabergoline therapy in vivo. All D2-expressing adenomas showed decreased cortisol levels in vivo in response to cabergoline therapy, whereas D2-negative cases did not. Preliminary data from another research group showed similar in vivo response rates after 20 months of cabergoline therapy [82]. In this study 3/8 CD patients (37.5%) had a complete normalization of 24 h UFC, 3/8 (37.5%) had a partial response (UFC ≤ 1.25 × ULN), whereas the remaining two patients did not respond. In both studies, cabergoline therapy was generally well tolerated. Despite the convincing results from both studies, the duration of treatment is still relatively short. For a genuine assessment of the utility of the drug cabergoline as a medical treatment for CD, studies on its efficacy and safety when used on a long-term basis in larger groups of patients are much needed. These studies are currently ongoing and some preliminary results indicate sustained efficacy after 1–2 years of follow-up [83].
One important issue that recently has dominated the field of medical therapy with DA-agonists has been the possible association between valvular heart disease and long-term therapy with the ergot-derived dopamine agonists (EDDA) pergolide and cabergoline. Two important papers were published in early 2007, which reported significantly increased risks (RR, 4.6–7.3) of valvular regurgitation in patients with idiopathic Parkinson’s disease that had received chronic treatment with either one of these drugs [84, 85]. Other studies have recently confirmed these data [86]. The pathogenetic mechanism behind this deleterious side effect is thought to be the binding of EDDA to 5-HT2B receptors expressed in the endocardial tissue of heart valves [85].
These findings have led to a number of important actions, including the withdrawal of pergolide from the US market. The impact of these studies on the (future) use of cabergoline in patients with CD cannot be fully determined yet, as one important issue needs to be emphasized. The maximum dose of cabergoline prescribed in CD is around 0.65 mg per day (4.5 mg/week), whereas the patients with Parkinson’s disease in the study by Zanettini et al. [85] received an average daily dose of 3.6 mg/day. In the other study by Schade et al. [84], an important risk difference was found between patients taking >3 mg cabergoline daily for more than 6 months (RR 50.3, 95% CI 6.6–381.4) compared to those who took less than 3 mg daily (RR 2.6; 95% CI 0.5–12.8). Therefore, these observations in Parkinson’s disease patients can not be directly extrapolated towards lower-dose cabergoline therapy in CD. Monitoring of cardiac function in CD patients on long-term cabergoline therapy does seem to be a prerequisite, however.
3.2.4 Combined treatment with SS and DA agonists in CD
Due to the reported presence of both sst and DA receptors in human corticotroph adenomas and the fact that both receptor types can inhibit ACTH production in vitro, the concept of a combination therapy with both SS-analogs and DA-agonists in CD seems to be a feasible approach [87]. These studies could be performed by co-treatment with individual SS-analogs and DA-agonists (pasireotide + cabergoline) or perhaps, in the near future, by administration of SS-DA-chimeric compounds such as BIM-23A760, which displays high affinity for sst2, D2 and to a lesser extent sst5 (see Table 1). If functional heterodimerization of these receptor subtypes occurs in vivo, as has already been shown to occur in vitro by different groups, this type of treatment could result in greatly enhanced efficacy of these compounds [29]. Also, as corticotroph adenomas can differ considerably in the total number of sst and D2 receptors they express [51, 52, 71], targeting of multiple receptors could increase the overall response rate in this group as a whole, compared to the use of individual SS or DA agonists. This has already been shown for GH-producing adenomas, where BIM-23A760 had overall superior efficacy compared to individual sst2, sst5 or D2-targeting agonists in terms of in vitro GH inhibition [88, 89]. As it is known that also in CD only subsets of patients have responded to either cabergoline or pasireotide monotherapy in vivo, it may well be that similar phenomena occur in corticotroph adenomas and that combination therapy can increase overall response rates. Until now, no studies have been published that have investigated this hypothesis.
Theoretically, co-treatment with sst and DA agonists may have other advantages as well. As stated before, the inefficacy of sst2-preferring compounds in CD, is probably due to down-regulation of sst2-expression by high levels of circulating glucocorticoids [48, 50, 51]. Inversely, if combined treatment with these analogs is effective and thus lowers cortisol levels in these patients, this could result in a return of sst2 expression. The latter would result in enhanced efficacy of SS-analogs with sst2-affinity and hence strongly increase pharmacotherapeutical options in these patients [59, 90].
4 Ectopic ACTH-producing syndrome (EAS)
4.1 Somatostatin analogs
For some decades it has been known that neuro-endocrine tumours that cause ectopic ACTH-producing syndrome (EAS), such as bronchial carcinoids or small cell lung cancer (SCLC), often express functional SS receptors. A number of smaller studies and case reports have been published on the use of Octreotide in patients with EAS. Interestingly, Octreotide was efficacious in lowering cortisol levels in a significant number of these patients, as opposed to the studies performed in patients with CD [91–94]. This discrepancy is further confirmed by the fact that many patients with EAS have positive lesions on 111In-pentereotide scan (Octreoscan), whereas most patients with CD do not [95]. The observation that many of the EAS producing neuro-endocrine tumours have functional sst2 receptors, despite the chronic hypercortisolism they are exposed to, could be explained by aberrant glucocorticoid receptor signalling in these tumour cells. This has been investigated extensively by a number of research groups over the past twenty years. It was found that many of the cell lines, derived from EAS producing small-cell lung carcinomas, carry gross mutations in the genetic sequence of the glucocorticoid receptor (GR) [96, 97]. These can be located either in the DNA-binding or the ligand-binding domain, but can also involve a number of transcription factors. The loss of function of the GR has important impact on POMC production in these cells. Any form of negative feedback is generally lost in these cells, leading to excessive and uninhibited production of POMC and ultimately, the full clinical spectrum of Cushing’s Syndrome. Another result of aberrant GR functioning, may be that glucocorticoid-induced down-regulation of somatostatin receptor subtype 2 (sst2) does not occur in these tumours, as opposed to pituitary-derived corticotroph adenomas. This could well explain the relatively high degree of positive Octreoscans and reported efficacy of Octreotide in this group of neuro-endocrine tumours [98, 99].
One main concern with the use of SS analogs in EAS, however, appears to be the long-term control of hypercortisolism. Although initial responses to Octreotide are frequent, these are not always sustained and treatment escapes are commonly encountered, due to a number of possible mechanisms of tachyphylaxis [100].
4.2 Dopamine agonists
Farrell et al. [69] showed in 1992 that the dopamine agonist bromocriptine could effectively inhibit POMC mRNA and ACTH precursor secretion in a small cell lung cancer cell line (CORL103), that is known to cause EAS. After these initial observations, to our knowledge no (clinical) studies have been performed that investigated the potential use of DA compounds in EAS, until a recent study by Pivonello et al. [101]. In this study, six patients with EAS-causing carcinoid tumours (four lung, one thymic, one pancreatic) underwent surgery. Five out of these 6 resected EAS tumours expressed D2, as determined by IHC. Three patients had persistent EAS after surgery and were treated with cabergoline at 3.5 mg/week for 6 months. All three patients had measurable D2 mRNA and two out of three had D4 mRNA expression on RT-PCR. Two patients had complete normalization of UFC after 3 months of cabergoline treatment, although one of them had a treatment escape afterwards. Of note, the long-term responder had the strongest overall D2 expression, including the D2 short isoform, and was also D4-receptor positive. In other pituitary tumours this expression profile has been associated with a good response to cabergoline therapy [71, 102]. Despite the small size of this study, it is probable that at least a subgroup of EAS patients could benefit from D2-targeted treatment, but obviously these results need to be confirmed in larger series.
4.3 Somatostatin–dopamine chimeras
Another interesting development in EAS tumours is the evaluation of dopamine-somatostatin chimeric molecules. Ferone et al. [30] examined the effects of the chimeras BIM-23A370 and BIM-23A387 in the non-small cell lung cancer cell line CALU-6. It was found that in these cells that natively express both sst2 and D2 receptors at high levels, the chimeric compounds had a stronger direct inhibitory effect on cell proliferation than the SS and DA-selective compounds alone. These data clearly confirm the hypothesis of heterodimerization between these two receptors, as had been suggested before by other groups [29].
A recent case report provides some further clinical evidence for this potential synergism between sst and DA receptors in EAS. In this case, a man with EAS due to a lung carcinoid tumour was treated medically after incomplete surgical removal. Cortisol levels normalized only temporarily with either SS-analog (Lanreotide) or dopamine agonist (Cabergoline) therapy alone. However, when both drugs were given simultaneously, based on co-expression of sst5 and D2 that was demonstrated by RT-PCR on the resected tumour specimen, the patient came into complete and prolonged remission [103].
5 Adrenal Cushing’s syndrome
Immunohistochemical and RT-PCR studies have shown that all sst subtypes (1–5) are expressed in the majority of normal adrenal cortices as well as in most cortisol-producing adenomas [104–106]. Interestingly, immunostaining occurred only in a minority (<30%) of these adenoma cells. One of the studies described a remarkably high expression of sst4 in the adenomas [104], but this was not found in another study [105]. To our knowledge, no studies have been performed so far to evaluate SS-analog therapy in cortisol-producing adenomas and carcinomas in vitro. In terms of clinical studies, octreotide does not appear to be effective in cases of adrenal carcinomas [107].
DA receptors (D1 and D2-like) are abundantly expressed in normal human adrenal cortex, where they are involved in the regulation of aldosterone release, but also in cortisol and androgen production [108, 109]. Their presence has been clearly established in different types of adrenal adenomas and carcinomas, some of which cause CS [110, 111]. D2 and D4 receptor subtypes were found in the cortisol-producing adrenal adenomas and carcinomas by RT-PCR. The adenomas expressed both D2 isoforms (short and long), whereas the carcinomas only expressed the D2-long isoform. Functional studies showed that the D2-agonist cabergoline was able to modulate ACTH- and angiotensin II-stimulated aldosterone release [111].
Taken together, despite the abundant expression of both SS and DA receptors in both the normal and the abnormal human adrenal gland, there are currently no data that would strongly support the use of SS-analog or DA-agonist therapy in patients with adrenal adenomas or carcinomas that cause CS. Given the high degree of surgical cure that can be obtained in most of these patients, the contribution of SS and DA-agonist therapy in this patient group is likely to be low.
6 Summary
In all forms of Cushing’s syndrome (pituitary, ectopic and adrenal CS) a subset of patients cannot be cured by surgery alone and therefore requires effective adjuvant treatment. Medical therapy can offer important advantages over other secondary treatments such as radiotherapy and bilateral adrenalectomy, including a relatively rapid onset of action and the fact that integrity of the HPA-axis is maintained. In CD, sst5 and D2 receptors play a crucial in the regulation of ACTH-release from corticotroph adenoma cells both in vitro and in vivo. Agents that selectively target these receptors can significantly reduce urinary cortisol excretion in subsets of patients. Combined treatment with these agents could in theory result in increased overall response rates, but these studies still need to be performed. In selected cases of ectopic ACTH-producing syndrome, D2-agonists are shown to be effective in reducing cortisol production, either alone or in combination with SS-analogs. Tumours that cause adrenal CS do express SS and DA receptors, but there is only limited data on the efficacy of agents that target these receptors neither in vitro nor in vivo. Their overall contribution to the management of these patients is likely to be low.
References
Orth DN. Cushing’s syndrome. N Engl J Med 1995;332(12):791–803.
Lindholm J, et al. Incidence and late prognosis of cushing’s syndrome: a population-based study. J Clin Endocrinol Metab 2001;86(1):117–23.
Newell-Price J, et al. Cushing’s syndrome. Lancet. 2006;367(9522):1605–17.
Atkinson AB, et al. Long-term remission rates after pituitary surgery for Cushing’s disease: the need for long-term surveillance. Clin Endocrinol 2005;63(5):549–59.
Benveniste RJ, et al. Repeated transsphenoidal surgery to treat recurrent or residual pituitary adenoma. J Neurosurg 2005;102(6):1004–12.
Locatelli M, Vance ML, Laws ER. Clinical review: the strategy of immediate reoperation for transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab 2005;90(9):5478–82.
Mahmoud-Ahmed AS, Suh JH. Radiation therapy for Cushing’s disease: a review. Pituitary 2002;5(3):175–80.
Vance ML. Pituitary radiotherapy. Endocrinol Metab Clin North Am 2005;34(2):479–87. xi.
Jagannathan J, et al. Gamma Knife surgery for Cushing’s disease. J Neurosurg 2007;106(6):980–7.
Ilias I, et al. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005;90(8):4955–62.
Isidori AM, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab 2006;91(2):371–7.
Morris D, Grossman A. The Medical management of Cushing’s syndrome. Ann NY Acad Sci 2002;970(1):119–33.
Castinetti F, et al. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol 2008;158(1):91–9.
Nieman LK. Medical therapy of Cushing’s disease. Pituitary 2002;5(2):77–82.
Barnett P. Somatostatin and somatostatin receptor physiology. Endocrine 2003;20(3):255–64.
Lamberts SW, Krenning EP, Reubi JC. The role of somatostatin and its analogs in the diagnosis and treatment of tumors. Endocr Rev 1991;12(4):450–82.
Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol 1999;20(3):157–98.
Missale C, et al. Dopamine receptors: from structure to function. Physiol Rev 1998;78(1):189–225.
Reubi JC, et al. Somatostatin receptors in human endocrine tumors. Cancer Res 1987;47(2):551–8.
Pivonello R, Ferone D, Lombardi G, Colao A, Lamberts SW, Hofland LJ. Novel insights in dopamine receptor physiology. Eur J Endocrinol 2007;156(Supplement 1):S13–S21.
Giustina G, et al. Dose-response study of the inhibiting effect of somatostatin on growth hormone and insulin secretion in normal subjects and acromegalic patients. Metabolism 1975;24(7):807–15.
Lamberts SW. The role of somatostatin in the regulation of anterior pituitary hormone secretion and the use of its analogs in the treatment of human pituitary tumors. Endocr Rev 1988;9(4):417–36.
Lamberts SW, et al. The somatostatin analog SMS 201–995 induces long-acting inhibition of growth hormone secretion without rebound hypersecretion in acromegalic patients. J Clin Endocrinol Metab 1985;60(6):1161–5.
Lamberts SW, et al. Long-term treatment of acromegaly with the somatostatin analogue SMS 201–995. N Engl J Med 1985;313(25):1576–80.
Saveanu A, et al. Bim-23244, a somatostatin receptor subtype 2- and 5-selective analog with enhanced efficacy in suppressing growth hormone (GH) from octreotide-resistant human GH-secreting adenomas. J Clin Endocrinol Metab 2001;86(1):140–5.
Bruns C, et al. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol 2002;146(5):707–16.
Molitch ME, et al. Bromocriptine as primary therapy for prolactin-secreting macroadenomas: results of a prospective multicenter study. J Clin Endocrinol Metab 1985;60(4):698–705.
Colao A, Lombardi G, Annunziato L. Cabergoline. Expert Opin Pharmacother 2000;1(3):555–74.
Rocheville M, et al. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 2000;288(5463):154–7.
Ferone D, et al. Somatostatin and dopamine receptor expression in lung carcinoma cells and effects of chimeric somatostatin–dopamine molecules on cell proliferation. Am J Physiol Endocrinol Metab 2005;289(6):E1044–1050.
Brazeau P, et al. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973;179(68):77–9.
O’Carroll AM, Krempels K. Widespread distribution of somatostatin receptor messenger ribonucleic acids in rat pituitary. Endocrinology 1995;136(11):5224–7.
Day R, et al. Expression of mRNA for somatostatin receptor (sstr) types 2 and 5 in individual rat pituitary cells. A double labeling in situ hybridization analysis. Endocrinology 1995;136(11):5232–5.
Mezey E, et al. Cell specific expression of the sst2A and sst5 somatostatin receptors in the rat anterior pituitary. Endocrinology 1998;139(1):414–9.
Brown MR, Rivier C, Vale W. Central nervous system regulation of adrenocorticotropin secretion: role of somatostatins. Endocrinology 1984;114(5):1546–9.
Kraicer J, Gajewski TC, Moor BC. Release of pro-opiomelanocortin-derived peptides from the pars intermedia and pars distalis of the rat pituitary: effect of corticotrophin-releasing factor and somatostatin. Neuroendocrinology 1985;41(5):363–73.
Lamberts SW, et al. Studies on the conditions determining the inhibitory effect of somatostatin on adrenocorticotropin, prolactin and thyrotropin release by cultured rat pituitary cells. Neuroendocrinology 1989;50(1):44–50.
Stafford PJ, et al. The pituitary-adrenal response to CRF-41 is unaltered by intravenous somatostatin in normal subjects. Clin Endocrinol (Oxf) 1989;30(6):661–6.
Hall R, et al. Action of growth-hormone-release inhibitory hormone in healthy men and in acromegaly. Lancet 1973;2(7829):581–4.
Lightman SL, Fox P, Dunne MJ. The effect of SMS 201–995, a long-acting somatostatin analogue, on anterior pituitary function in healthy male volunteers. Scand J Gastroenterol Suppl 1986;119:84–95.
Invitti C, et al. Effect of sandostatin on CRF-stimulated secretion of ACTH, beta-lipotropin and beta-endorphin. Horm Metab Res 1991;23(5):233–5.
Fehm HL, et al. Somatostatin: a potent inhibitor of ACTH-hypersecretion in adrenal insufficiency. Klin Wochenschr 1976;54(4):173–5.
Cervia D, et al. Pharmacological characterisation of native somatostatin receptors in AtT-20 mouse tumour corticotrophs. Br J Pharmacol 2003;139(1):109–21.
Richardson UI, Schonbrunn A. Inhibition of adrenocorticotropin secretion by somatostatin in pituitary cells in culture. Endocrinology 1981;108(1):281–90.
Tallent M, et al. Somatostatin receptor subtypes SSTR2 and SSTR5 couple negatively to an L-type Ca2 + current in the pituitary cell line AtT-20. Neuroscience 1996;71(4):1073–81.
Strowski MZ, et al. Somatostatin receptor subtypes 2 and 5 inhibit corticotropin-releasing hormone-stimulated adrenocorticotropin secretion from AtT-20 cells. Neuroendocrinology 2002;75(6):339–46.
Cervia D, Fehlmann D, Hoyer D. Native somatostatin sst2 and sst5 receptors functionally coupled to Gi/o-protein, but not to the serum response element in AtT-20 mouse tumour corticotrophs. Naunyn Schmiedebergs Arch Pharmacol 2003;367(6):578–87.
van der Hoek J, et al. Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am J Physiol Endocrinol Metab 2005;289(2):E278–287.
Schonbrunn A. Glucocorticoids down-regulate somatostatin receptors on pituitary cells in culture. Endocrinology 1982;110(4):1147–54.
Stalla GK, et al. Octreotide exerts different effects in vivo and in vitro in Cushing’s disease. Eur J Endocrinol 1994;130(2):125–31.
Hofland LJ, et al. The multi-ligand somatostatin analogue SOM230 inhibits ACTH secretion by cultured human corticotroph adenomas via somatostatin receptor type 5. Eur J Endocrinol 2005;152(4):645–54.
Batista DL, et al. The effects of SOM230 on cell proliferation and adrenocorticotropin secretion in human corticotroph pituitary adenomas. J Clin Endocrinol Metab 2006;91(11):4482–8.
Danila DC, et al. Somatostatin Receptor-Specific Analogs: Effects on Cell Proliferation and Growth Hormone Secretion in Human Somatotroph Tumors 10.1210/jc.86.7.2976. J Clin Endocrinol Metab 2001;86(7):2976–81.
Lamberts SW, Uitterlinden P, Klijn JM. The effect of the long-acting somatostatin analogue SMS 201–995 on ACTH secretion in Nelson’s syndrome and Cushing’s disease. Acta Endocrinol (Copenh) 1989;120(6):760–6.
Ambrosi B, et al. Failure of somatostatin and octreotide to acutely affect the hypothalamic-pituitary-adrenal function in patients with corticotropin hypersecretion. J Endocrinol Invest 1990;13(3):257–61.
Tyrrell JB, et al. Inhibition by somatostatin of ACTH secretion in Nelson’s syndrome. J Clin Endocrinol Metab 1975;40(6):1125–7.
Petrini L, et al. Long-term treatment of Nelson’s syndrome by octreotide: a case report. J Endocrinol Invest 1994;17(2):135–9.
Kelestimur F, et al. The effects of octreotide in a patient with Nelson’s syndrome. Postgrad Med J 1996;72(843):53–4.
van der Hoek J, Lamberts SW, Hofland LJ. The role of somatostatin analogs in Cushing’s disease. Pituitary 2004;7(4):257–64.
Boscaro M, Petersenn S, Atkinson AB, Bertherat J, Findling J, Snyder P, McBride K, Reincke M, Ludlam W, Gao B, Melmed S, Freda P, Frohman L, Grossman A, Biller B, Glusman JE. Pasireotide (SOM230), the novel multi-ligand somatostatin analogue, is a promising medical therapy for patients with Cushing’s disease: preliminary safety and efficacy results of a phase II study. Presented at ENDO 2006, abstr OR9-1, 2006,. Boston, USA.
Ma P, et al. Pharmacokinetic-pharmacodynamic comparison of a novel multiligand somatostatin analog, SOM230, with octreotide in patients with acromegaly. Clin Pharmacol Ther 2005;78(1):69–80.
Weckbecker G, et al. SOM230: a new somatostatin peptidomimetic with potent inhibitory effects on the growth hormone/insulin-like growth factor-I axis in rats, primates, and dogs. Endocrinology 2002;143(10):4123–30.
Antakly T, et al. Induced expression of the glucocorticoid receptor in the rat intermediate pituitary lobe. Science 1985;229(4710):277–9.
Stack J, Surprenant A. Dopamine actions on calcium currents, potassium currents and hormone release in rat melanotrophs. J Physiol 1991;439:37–58.
Farah JM Jr., Malcolm DS, Mueller GP. Dopaminergic inhibition of pituitary beta-endorphin-like immunoreactivity secretion in the rat. Endocrinology 1982;110(2):657–9.
Saiardi A, Borrelli E. Absence of dopaminergic control on melanotrophs leads to Cushing’s-like syndrome in mice. Mol Endocrinol 1998;12(8):1133–9.
Lamberts SW, de Lange SA, Stefanko SZ. Adrenocorticotropin-secreting pituitary adenomas originate from the anterior or the intermediate lobe in Cushing’s disease: differences in the regulation of hormone secretion. J Clin Endocrinol Metab 1982;54(2):286–91.
Croughs RJ, et al. Bromocriptine-responsive Cushing’s disease associated with anterior pituitary corticotroph hyperplasia or normal pituitary gland. J Clin Endocrinol Metab 1989;68(2):495–8.
Farrell WE, et al. Bromocriptine inhibits pro-opiomelanocortin mRNA and ACTH precursor secretion in small cell lung cancer cell lines. J Clin Invest 1992;90(3):705–10.
Yin D, et al. Induction of apoptosis in murine ACTH-secreting pituitary adenoma cells by bromocriptine. FEBS Lett 1994;339(1–2):73–5.
Pivonello R, et al. Dopamine receptor expression and function in corticotroph pituitary tumors. J Clin Endocrinol Metab 2004;89(5):2452–62.
Stefaneanu L, et al. Dopamine D2 receptor gene expression in human adenohypophysial adenomas. Endocrine 2001;14(3):329–36.
Miller JW, Crapo L. The medical treatment of Cushing’s syndrome. Endocr Rev 1993;14(4):443–58.
Lamberts SW, et al. The mechanism of the suppressive action of bromocriptine on adrenocorticotropin secretion in patients with Cushing’s disease and Nelson’s syndrome. J Clin Endocrinol Metab 1980;51(2):307–11.
Pivonello R, et al. Complete remission of Nelson’s syndrome after 1-year treatment with cabergoline. J Endocrinol Invest. 1999;22(11):860–5.
Petrossians P, et al. ACTH silent adenoma shrinking under cabergoline. Eur J Endocrinol 2001;144(1):51–7.
Miyoshi T, et al. Effect of cabergoline treatment on Cushing’s disease caused by aberrant adrenocorticotropin-secreting macroadenoma. J Endocrinol Invest. 2004;27(11):1055–9.
Casulari LA, et al. Nelson’s syndrome: complete remission with cabergoline but not with bromocriptine or cyproheptadine treatment. Horm Res. 2004;62(6):300–5.
Shraga-Slutzky I, Shimon I, Weinshtein R. Clinical and biochemical stabilization of Nelson’s syndrome with long-term low-dose cabergoline treatment. Pituitary. 2006;9(2):151–4.
Illouz F, et al. [Use of cabergoline in persisting Cushing’s disease]. Ann Endocrinol (Paris) 2006;67(4):353–6.
Garcia C, et al. [Nelson’s syndrome management: current knowledge]. Rev Med Interne. 2007;28(11):766–9.
Godbout A, B.H., Babin S, Sabourin A, Lacroix A. Cabergoline in the long-term treatment of Cushing’s disease. in The Endocrine Society’s 89th Annual Meeting, June 2–5. 2007, Toronto, Canada.
Pivonello R, D.M.M., Faggiano A, De Leo M, Lombardi G, Hofland L, Lamberts S, Colao A. Cabergoline treatment in Cushing’s disease: Effect on hypertension, glucose intolerance and dyslipidemia. in P4–50, Endocrine Society’s 89th Annual Meeting, June 2–5. 2007, Toronto, Canada.
Schade R, et al. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med 2007;356(1):29–38.
Zanettini R, et al. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med 2007;356(1):39–46.
Rasmussen VG, et al. Heart valve disease associated with treatment with ergot-derived dopamine agonists: a clinical and echocardiographic study of patients with Parkinson’s disease. J Intern Med 2008;263(1):90–8.
Colao A, et al. Combined therapy of somatostatin analogues and dopamine agonists in the treatment of pituitary tumours. Eur J Endocrinol 2007;156(Suppl 1):S57–63.
Saveanu A, et al. Somatostatin and dopamine-somatostatin multiple ligands directed towards somatostatin and dopamine receptors in pituitary adenomas. Neuroendocrinology 2006;83(3–4):258–63.
Jaquet P, et al. BIM-23A760, a chimeric molecule directed towards somatostatin and dopamine receptors, vs universal somatostatin receptors ligands in GH-secreting pituitary adenomas partial responders to octreotide. J Endocrinol Invest 2005;28(11 Suppl International):21–7.
Schmid HA. Pasireotide (SOM230): Development, mechanism of action and potential applications. Mol Cell Endocrinol. 2008;286:69–74.
Lamberts SW, et al. A role of (labeled) somatostatin analogs in the differential diagnosis and treatment of Cushing’s syndrome. J Clin Endocrinol Metab 1994;78(1):17–9.
Phlipponneau M, et al. Somatostatin analogs for the localization and preoperative treatment of an adrenocorticotropin-secreting bronchial carcinoid tumor. J Clin Endocrinol Metab 1994;78(1):20–4.
Bertagna X, et al. Suppression of ectopic adrenocorticotropin secretion by the long-acting somatostatin analog octreotide. J Clin Endocrinol Metab 1989;68(5):988–91.
Vignati F, Loli P. Additive effect of ketoconazole and octreotide in the treatment of severe adrenocorticotropin-dependent hypercortisolism. J Clin Endocrinol Metab 1996;81(8):2885–90.
de Herder WW, et al. Somatostatin receptor scintigraphy: its value in tumor localization in patients with Cushing’s syndrome caused by ectopic corticotropin or corticotropin-releasing hormone secretion. Am J Med 1994;96(4):305–12.
Ray DW, et al. Human small cell lung cancer cell lines expressing the proopiomelanocortin gene have aberrant glucocorticoid receptor function. J Clin Invest 1994;93(4):1625–30.
Gaitan D, et al. Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Mol Endocrinol 1995;9(9):1193–201.
Uwaifo GI, et al. Is there a therapeutic role for octreotide in patients with ectopic Cushing’s syndrome? J Endocrinol Invest 2003;26(8):710–7.
Lamberts SW, et al. Successful treatment with SMS 201-995 of Cushing’s syndrome caused by ectopic adrenocorticotropin secretion from a metastatic gastrin-secreting pancreatic islet cell carcinoma. J Clin Endocrinol Metab 1988;67(5):1080–3.
Hofland LJ, Lamberts SW. The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr Rev 2003;24(1):28–47.
Pivonello R, et al. Dopamine receptor expression and function in corticotroph ectopic tumors. J Clin Endocrinol Metab 2007;92(1):65–9.
Pivonello R, et al. Dopamine receptor expression and function in clinically nonfunctioning pituitary tumors: comparison with the effectiveness of cabergoline treatment. J Clin Endocrinol Metab 2004;89(4):1674–83.
Pivonello R. Cabergoline plus lanreotide for ectopic Cushing’s syndrome. N Engl J Med 2005;352(23):2457–8.
Unger N, et al. Immunohistochemical localization of somatostatin receptor subtypes in benign and malignant adrenal tumors. Clin Endocrinol (Oxf) 2008;68:850–7.
Ueberberg B, et al. Differential expression of the human somatostatin receptor subtypes sst1 to sst5 in various adrenal tumors and normal adrenal gland. Horm Metab Res 2005;37(12):722–8.
Unger N, et al. Immunohistochemical determination of somatostatin receptor subtypes 1, 2A, 3, 4, and 5 in various adrenal tumors. Endocr Res 2004;30(4):931–4.
de Herder WW, Lamberts SW. Is there a role for somatostatin and its analogs in Cushing’s syndrome? Metabolism 1996;45(8 Suppl 1):83–5.
Missale C, et al. Dopaminergic receptor mechanisms modulating the renin-angiotensin system and aldosterone secretion: an overview. J Cardiovasc Pharmacol 1989;14(Suppl 8):S29–39.
Amenta F, et al. Pharmacological characterization and autoradiographic localization of dopamine receptors in the human adrenal cortex. Eur J Endocrinol 1994;131(1):91–6.
Wu KD, et al. Expression and localization of human dopamine D2 and D4 receptor mRNA in the adrenal gland, aldosterone-producing adenoma, and pheochromocytoma. J Clin Endocrinol Metab 2001;86(9):4460–7.
Pivonello R, et al. Dopamine receptor expression and function in human normal adrenal gland and adrenal tumors. J Clin Endocrinol Metab 2004;89(9):4493–502.
Schmid HA, Schoeffter P. Functional activity of the multiligand analog SOM230 at human recombinant somatostatin receptor subtypes supports its usefulness in neuroendocrine tumors. Neuroendocrinology 2004;80(Suppl 1):47–50.
Newman-Tancredi A, et al. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor. J Pharmacol Exp Ther 2002;303(2):805–14.
Jaquet P, et al. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur J Endocrinol 2005;153(1):135–41.
Hofland LJ, et al. The novel somatostatin analog SOM230 is a potent inhibitor of hormone release by growth hormone- and prolactin-secreting pituitary adenomas in vitro. J Clin Endocrinol Metab 2004;89(4):1577–85.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure statement: The authors of this manuscript have nothing to disclose.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
de Bruin, C., Feelders, R.A., Lamberts, S.W.J. et al. Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s Syndrome. Rev Endocr Metab Disord 10, 91–102 (2009). https://doi.org/10.1007/s11154-008-9082-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-008-9082-4