
Abstract
Chlorophyll (Chl) b serves an essential function in accumulation of light-harvesting complexes (LHCs) in plants. In this article, this role of Chl b is explored by considering the properties of Chls and the ligands with which they interact in the complexes. The overall properties of the Chls, not only their spectral features, are altered as consequences of chemical modifications on the periphery of the molecules. Important modifications are introduction of oxygen atoms at specific locations and reduction or desaturation of sidechains. These modifications influence formation of coordination bonds by which the central Mg atom, the Lewis acid, of Chl molecules interacts with amino acid sidechains, as the Lewis base, in proteins. Chl a is a versatile Lewis acid and interacts principally with imidazole groups but also with sidechain amides and water. The 7-formyl group on Chl b withdraws electron density toward the periphery of the molecule and consequently the positive Mg is less shielded by the molecular electron cloud than in Chl a. Chl b thus tends to form electrostatic bonds with Lewis bases with a fixed dipole, such as water and, in particular, peptide backbone carbonyl groups. The coordination bonds are enhanced by H-bonds between the protein and the 7-formyl group. These additional strong interactions with Chl b are necessary to achieve assembly of stable LHCs.
Similar content being viewed by others
Introduction
The dramatic developmental transformation performed by the chloroplast has attracted broad interest over the past several decades (see Hoober and Argyroudi-Akoyunoglou 2004; Wise and Hoober 2006, for reviews). Although the organelle displays a variety of features among different organisms, as revealed by electron microscopy, its monophyletic origin by endosymbiosis of an ancient cyanobacterium has received increasingly strong support (Palmer 2003; Bhattacharya and Medlin 2004; Rodriguez-Ezpeleta et al. 2005). Descendents of the primary endosymbiotic event branched into the glaucophytes, green algae, and plants, which contain chlorophyll (Chl) a and Chl b, and the red algae, which contain only Chl a. Tomitani et al. (1999) provided evidence that the genes for chlorophyllide a oxygenase (CAO), the enzyme that catalyzes conversion of chlorophyllide (Chlide) a to Chlide b, also have a common origin. The evolutionary relationship of CAO in plants with the enzyme in the cyanobacterial prochlorophytes indicates that the original ancestor of plastids contained Chl b and that modern cyanobacteria, along with the red algae, lost this ability. Divergence from a secondary endosymbiotic event, also apparently singular, of a red alga gave rise to four major groups of chromophyte algae, the dinoflagellates, heterokonts, haptophytes, and cryptophytes, that contain Chl c as a major pigment in addition to Chl a (Bachvaroff et al. 2005; Shalchian-Tabrizi et al. 2006). This lineage suggests that the same fundamental mechanisms should underlie processes in chloroplast development in all plant species. Among these are expected to be the mechanisms that guide the interactions of Chls, proteins, and lipids during assembly of the thylakoid membrane. Even with the extensive studies already done on these processes, our understanding of many details of these mechanisms remains clouded.
This article will consider physicochemical factors that are likely fundamental in the assembly of light-harvesting complexes (LHCs) in the plastids of eukaryotic organisms. Particular emphasis is given to the properties of Chls a, b, and c and their interactions with ligands. Chls b and c occur essentially exclusively in LHCs. Whereas Chl a is ubiquitous, it alone is not sufficient for LHC assembly. Chl c seems to play the same role in LHC assembly in chromophyte algae as Chl b does in green algae and plants (Durnford et al. 1999; De Martino et al. 2000; Goss et al. 2000). Thus a principal question is the step in LHC assembly for which synthesis of Chl b is required. It is likely that the plastids derived from the secondary endosymbiotic event solved this problem by finding a pathway to Chl c. The Chl-binding proteins in these organisms are evolutionarily related to those in green algae and plants (Schmitt et al. 1994; Green and Durnford 1996). The relatively simple LHCs are well-defined structures and consequently are excellent systems to search for basic mechanisms. It is useful for illustration purposes to include Chl d, the most recently characterized member of the Chl family (Miyashita et al. 1997; Akiyama et al. 2002) found as the major Chl in the cyanobacterium Acaryochloris marina, which functions in core complexes in an analogous fashion to Chl a. Bacteriochlorophyll (BChl) a is also briefly mentioned for comparison.
Summary of differences in chlorophylls
The spectral differences between the species of Chls in chloroplasts expand the ability of photosynthetic organisms to harvest light. However, the spectral range of Chl a alone is broadened by various micro-environments within complexes with proteins (Nishigaki et al. 2001; Croce et al. 2002; Linnanto et al. 2006), which argues against spectral differences per se as the raison d’être for occurrence of the “secondary” Chls, in particular, Chls b and c. Overall properties, not only their spectral features, are altered as consequences of chemical modifications on the periphery of the molecules. Important modifications are desaturation of sidechains and introduction of oxygen atoms at specific locations. Oxygen is the most electronegative atom commonly found in biological systems and exerts significant effects on the electronic distribution in the Chl molecule.
We previously (Hoober and Eggink 1999; Eggink et al. 2001, 2004; Chen et al. 2005) proposed that modifications at the periphery of the Chl molecule influence the coordination chemistry of the central Mg atom and that this effect plays an important, if not major, role in the interaction of Chl b with LHC apoproteins (LHCPs) and thus in the assembly of LHCs. In particular, we proposed that, as a Lewis acid, the Mg atom in Chl b favors axial coordination bonds with harder Lewis bases than does Chl a. This proposal was supported experimentally by direct measurement of equilibrium constants of various tetrapyrrole derivatives with specific ligands (Tamiaki et al. 1998). In an unbiased chemical context, Chls a and b should then prefer different ligands. Recent evidence demonstrated, however, that Chl a interacts with a broad range of ligands, from the imidazole group of histidine to water. Chl b, on the other hand, is found only with ligands containing an oxygen atom.
Chlorophylls a and d
Conversion of 3,8-divinyl-Chl a to 3-monovinyl-Chl a by reduction of the 8-vinyl group to an ethyl group is the final step in Chl a biosynthesis and yields the predominant form of Chl a (Nagata et al. 2005). As a result, Chl a has electron-donating methyl and ethyl groups at positions 7 and 8, respectively (Fig. 1). Along with reduction of the C17–C18 double bond to a single bond, which converts the porphyrin precursor protochlorophyllide (Pchlide) to the chlorin ring system, these groups impose an electron density, from opposite sides of the molecule along the X axis, on the pyrrole nitrogens, which partially shields the positive charge of the central Mg atom. In addition, the 3-vinyl and 131-keto groups exert weak electron withdrawing effects on opposite ends of the Y axis.

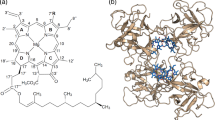
Structures of the major Chls. Except for the oxidation of the 7-methyl group in Chl a to the formyl group in Chl b, Chls a and b are identical. Chl d contains a formyl group at position 3. Chls a, b, and d include the 20-carbon isoprene alcohol, phytol (Ph), esterified to the carboxyl group at position 173. This carboxyl group remains unesterified in Chl c, which also contains double bonds in the sidechain between positions 171 and 172 and in the macrocycle between carbons 17 and 18. These additional double bonds extend conjugation of the macrocyclic π system to the free carboxyl group. Chl c species differ at positions 7 and 8; c 1: 7, –CH3, 8, –C2H5; c 2: 7, –CH3, 8, –C2H3; c 3: 7, –COOCH3, 8, –C2H3 (shown in figure)
The geometrical coordinates of the molecular framework for Chl a are shown in Fig. 1. The primary X axis transects the molecule from the position of C17 to C7. The Y axis transects the molecule from C2 to C12. The experimentally determined, functional Q y transition-moment direction is a vector 70° clockwise from the X axis (Fragata et al. 1988; Simonetto et al. 1999; Sundholm 2003; Cai et al. 2006). This displacement from the geometrical Y axis places the functional Q y vector from near C1 to near C11. The functional Q x vector is within a few degrees of the geometrical X axis.
Chl d is synthesized by oxidation of the 3-vinyl group to a formyl group, whose electron-withdrawing character further extends the Q y vector but should also cause the transition direction to align more closely with the molecular Y axis. As a result, the lowest energy absorption band shifts from 665 nm (in methanol) for Chl a to a longer wavelength (lower energy) maximum of 697 nm for Chl d and increases the dipole strength, which is proportional to the molar absorption coefficient. The transition dipole is strengthened further in BChl a, which has an electron-withdrawing acetyl group on C3 and a single bond between C7 and C8. The Q y absorption maximum is shifted to 772 nm (in methanol), with a dipole of 7.2 Debye (D) (at a refractive index for the environment of 1.35) (Knox and Spring 2003). The electronic distribution in BChl a is more symmetrically aligned along the Y axis, with an elliptical electron density, and the Q y transition-moment direction is essentially perpendicular to the X axis, i.e., from C2 to C12 (Sundholm 2003).
Chlorophyll b
Synthesis of Chl b involves incorporation of the electronegative oxygen atom to generate the 7-formyl group, which, as an aromatic aldehyde, is expected to have a dipole moment for the group of approximately 3.0 D (Desyatnyk et al. 2005). The oxygen provides a significant pull on electrons away from the core of the molecule along the X axis, which weakens the Q y dipole strength of the molecule from 5.33 D for Chl a to 4.41 D in Chl b (at a refractive index for the environment of 1.35) (Knox and Spring 2003). Since the dipole strength determines the magnitude of the absorption coefficients, among other properties, the absorption coefficient of Chl b is only 50–62% (depending on solvent) of the Q y absorption peak of Chl a and is shifted to higher energy, with a maximum at 652 nm (in methanol). The Q y transition-moment direction is displaced further from the Y axis than in Chl a and is at an angle of only 61° clockwise from the X axis (Simonetto et al. 1999), described as a transect from near C20 to near C10.
Intuitively, the Q x transition moment of Chl b should be stronger than that of Chl a. Computational analysis of molecular orbitals suggest that the Q x oscillator strength of Chl b is 2-fold greater than for Chl a, with a maximum at 538 nm (Linnanto and Korppi-Tommola 2004). The magnetic circular dichroism (MCD) spectrum of Chl b contains a weak negative transition at 540 nm and an intense negative transition at 625 nm (Frackowiak et al. 1987). From the argument that a stronger molecular dipole is consistent with a longer wavelength absorption maximum, the longer-wavelength negative transition in the MCD spectrum of Chl b may correspond to the Q x transition.
The molecular orbital calculations suggest that, with weaker Q y and stronger Q x transitions, the electronic distribution in the Chl b molecule is essentially circular (Hoff and Amesz 1991). The lessened electron density around the pyrrole nitrogen atoms lowers the pK values for these atoms by about two pH units (Phillips 1963; Smith KM 1975). The withdrawal of electron density from the pyrrole nitrogens results in less shielding of the Mg atom of Chl b and allows it to more strongly express its positive point charge. Molecular orbital calculations give the Mg in Chl an atomic charge with a value of +0.7 to +1.3 (Linnanto and Korppi-Tommola 2004).
Chlorophyll c
Chl c is found with Chl a in chromophyte algae, where it functions as a light-harvesting pigment. Whereas most of these algae do not contain Chl b, some Prasinophycean algae, such as Montoniella squamata, accumulate a Chl c, 3,8-divinyl-Pchlide, along with Chls a and b (Schmitt et al. 1994; Green and Durnford 1996). The red alga that contributed its plastid in the secondary endosymbiotic event apparently lacked Chl c. Ability to make Chl c was the solution these organisms found to solve the problem that was accomplished in chlorophytes with Chl b. As shown in Fig. 1, the structure of Chl c 3 has several unique features. The c-type Chls characteristically retain the C17–C18 double bond that occurs in the porphyrin precursor, Pchlide a. In addition, a trans double bond is introduced between C171 and C172 in the sidechain, which extends conjugation of the ring π system to the usually unesterified, electronegative C173 carboxyl group. Conversion of the propionate sidechain of Pchlide a to the acrylate sidechain of Chl c inhibits the ability of NADPH:Pchlide oxidoreductase to reduce the C17–C18 double bond as occurs in Chl a synthesis (Helfrich et al. 2003), which suggests that sidechain desaturation occurs prior to potential interaction of Pchlide a with the oxidoreductase. Thus Chl c retains the porphyrin ring system. The three major sub-types of Chl c (c 1, c 2 and c 3) occur as the result of oxidation of the C7 and/or C8 substituents. The C7 methyl group is modified to a methylcarboxylate (–COOCH3) in Chl c 3 and the C8 vinyl group remains unreduced (Porra 1997). These remarkable modifications all lie on the X axis of the molecule. Absorbance spectra of the series (c 1, c 2 and c 3) suggest that the functional Q x transition moment increasingly dominates the long-wavelength absorption peaks (Jeffrey and Wright 1987; Helfrich et al. 2003), further reduces the absorption coefficient and blue-shifts the Q y absorbance maximum to 630 nm for Chl c 1 (in acetone). Additional modifications, such as esterification of the acrylate sidechain with galactosyl diacylglycerol, increase the number of minor forms of Chl c (Garrido et al. 2000).
A summary of ligands of chlorophylls in light-harvesting complexes
Coordination bonds are formed between Lewis acids and bases. A Lewis acid has an unfilled orbital that can accept a pair of electrons. A Lewis base (ligand) has a pair of unshared electrons that are available for donation to the Lewis acid to form a donor-acceptor complex. Lewis acids and bases are characterized as “soft” or “hard” according to their chemical properties (Jensen 1978). Soft species tend to bond by short-range orbital interactions, while hard species interact primarily by electrostatic forces.
The central Mg atom of Chl molecules, as the Lewis acid, interacts with proteins by formation of coordination bonds with an amino acid sidechain as the Lewis base. Compression of the electron cloud toward the Y axis of the Chl a molecule, as when the C17–C18 double-bond and C8 vinyl group are reduced, tends to shield the Mg atom and effectively reduces the electronegativity of the metal. This results in weaker interaction with the negative end of a fixed dipole or even repulsion of negatively charged groups. In contrast, in Chl b the Mg atom is less shielded and more strongly expresses its positive charge. Electron density in Chl b is also pulled outward by H-bonds between the 7-formyl group and other structures, which further enhances the Q x transition moment. Substituting the central Mg in BChl a with other metals (e.g., Ni) of greater electronegativity strongly influences the Q x but not the Q y transition energies of the tetrapyrrole molecule (Hartwich et al. 1998). In Chls a and b the metal is the same, but the argument can be applied in reverse, in which substitution of peripheral groups on the Q x axis alters the environment of the central Mg ion and thus its effective electronegativity.
If H+ is considered as a Lewis acid, the availability of electrons in a Lewis base should be reflected in its pK value (Jensen 1978) (pK values vary dramatically from those obtained in aqueous media when the ionizable group is located in a nonpolar micro-environment (Mehler er al. 2002)). The electron pair available on an amine nitrogen atom binds H+ strongly (pK ∼9). Although the nonpolar micro-environment within a protein molecule may lower the pK of an amino group of lysine (Gunner et al. 2000, 2006), the amine is usually protonated under physiological conditions, thus positively charged, and the electron pair is not available for coordination with Chl. Lysine amino groups are not ligands in membrane-spanning regions of Chl-binding proteins (Balaban et al. 2002; Ferreira et al. 2004; Liu et al. 2004). The strength of other amino acid sidechains as Lewis bases should then decrease in the order of decreasing pK, e.g., imidazole (pK 7) > carboxylate (pK 4–5) > peptide backbone amide (pK −0.42) > sidechain amide (pK −0.62) > water (pK −1.74). However, formation of Chl–ligand complexes does not follow this pattern. Chl a interacts as expected with imidazole, its predominant partner, but also with amide sidechains and water. Chl b does not interact significantly with imidazole, whereas the peptide backbone carbonyl group and water are favorable ligands. Orbital interactions may play a greater role in coordination bonds with ligands containing accessible electron pairs (i.e., high pK values) such as imidazole, whereas electrostatic interactions are more likely with ligands having a low pK value such as carbonyl groups. This comparison suggests that interactions of the Chls with ligands involve more than simply the availability of non-bonded electrons on the ligand. In addition to differences in the properties of the Lewis bases, the more exposed positively charged Mg ion in Chl b is more electronegative and acts as a harder Lewis acid than the metal in Chl a. Thus Chl b should favor electrostatic bonds with groups containing a strong, fixed dipole.
With the exception of the imidazole group, ligands to Chl contain oxygen. Lewis bases that contain oxygen atoms are polar, with a “fixed” dipole, and tend to be “hard” Lewis bases. The structures listed in Table 1, which are common or potential ligands of Chl, are ordered according to increasing dipole moment of the monomeric molecule, as calculated by ab initio methods, to emphasize the importance of this parameter. When these molecules interact by H-bonding with other molecules, as in a solution or a crystal structure, the dipole moments increase (Spackman 1992; Abramov et al. 1999; Whitfield et al. 2006). This effect is unlikely to be significant when ligands are isolated within the nonpolar environment in a membrane, although the dipole of a “polarizable” ligand is affected by its interaction with the Mg of Chl. The dipole moment of an alcohol is too weak to effectively compete with water and thus hydroxyl groups are not common ligands. Each productive ligand is discussed in more detail in the following.
Water
Water is the Lewis base that seems to be a “regulatory” ligand because of its strong interaction with Chl b and its weaker interaction with Chl a (Ballschmitter et al. 1969). In solution, where water is fully H-bonded (dielectric constant, 81), its dipole moment is 2.70 D (Table 1); in ice, this value is 3.09 D (Batista et al. 1998). In an environment in which the dielectric constant is 2–4, as occurs in a protein or membrane, the dipole of a water molecule is likely nearer to that in the gas phase, 1.85 D (Dyke and Muenter 1973). However, when associated with a positive charge such as the Mg in Chl, the dipole moment is probably near the H-bonded value. The charge at the negative end of the dipole of water provides an electrostatic contribution to the interaction.
For a functional group in a protein to form a coordination bond with the Mg atom in Chl, a water ligand, which is likely present throughout the latter steps in the biosynthetic pathway from Mg-protoporphyrin IX onward, must be displaced. It is interesting that three Chl b molecules retain water as a ligand and connect with the protein via a water bridge (see below).
Imidazole
The imidazole sidechain of histidine in the unprotonated form has an unshared pair of electrons on N(3) (designated as Nε2 by Standfuss et al. 2005). H+ binds to the electron pair with a pK value that lies within the range of 5–8, depending upon the environment. Nonpolar environments stabilize the unprotonated form, and thus the electron-rich imidazole group is available for coordination with the Mg of Chl a within a membrane. The dipole moment for imidazole is between 3.66 D (gas phase) and 4.80 D (crystal structure), with the predominant contribution to the dipole provided by the N(1)-H bond (Spackman 1992). When the N(1) hydrogen is replaced with the electron-donating methyl group, the resulting coordination bond at N(3) is stronger (van Gammeren et al. 2004). In aqueous solution, the dipole moment is enhanced to a value of 3.96 D by H-bonding (Table 1). Both N atoms have a small negative charge, and the electron density is distributed nearly symmetrically (Fig. 2). The aromatic character of imidazole allows the dipole to reorganize in response to interaction with another structure.

(A) The structure of the imidazole group of histidine and (B) its electronic charge density, determined by X-ray diffraction at 103 K for the projection in (A) (adapted from Epstein et al. 1982). In (A), R = remainder of the histidine molecule
His120, at the lumenal end of helix-2 in LHCII (see Fig. 3), is not a ligand to Chl a, possibly because of its exposure to the thylakoid lumen where competition with water is greater than within the membrane. Also, the imidazole group may be protonated at the pH of the lumen during active photosynthesis, estimated to be near pH 5 (Kramer et al. 1999; Sacksteder et al. 2000). His212, a ligand to Chl a, is also near the lumenal surface of the membrane but is likely shielded from the aqueous lumen by helix-4 of the LHC protein.

Model of the association of Chls with Lhcb1. The arrangement of the protein in thylakoid membranes is illustrated according to Green and Durnford (1996). The symbols designating the chlorin rings of the six Chl b molecules are filled (green). A water ligand for four Chl molecules is indicated by a central blue dot. The Chl a molecules are numbered 1–8 and the Chl b molecules 9–14, as designated by Standfuss et al. (2005)
The imidazole group provides a good example of the attractive/repulsive forces that limit the strength of the coordination bond. A repulsive force should exist between the π clouds of Chl and the ligand. However, the pair of electrons on N3 is an attractive force that satisfies the needs of the Lewis acid, Mg. The concept of electronegativity equalization (Noy et al. 2000) suggests that a partial charge (about 0.3e) is transferred from imidazole to the metal ion upon coordination (analyzed with Ni-BChl a). This shift in electron density should reduce the role of the dipole in the coordination bond and generate a partial positive charge on the ligand that is accommodated by the electron cloud of the conjugated π system of Chl a. In contrast, the more exposed positive charge on Mg in Chl b likely repels the positive charge that develops on the ligand. Thus the attractive force is limited by the repulsive force created between Mg and the induced positive charge on the ligand.
Carboxyl group
Two ligands for Chl a in LHCII are charge-compensated ion-pairs formed by electrostatic interaction between the sidechain carboxylate of glutamic acid and the guanidinium group of arginine. Although at pH 7 the carboxyl group has a negative charge and multiple pairs of unbonded electrons, these electrons are distributed between the two electronegative oxygen atoms in a resonance structure and are less available for protonation than in the imidazole group. This property is reflected in the higher-proton concentration (pH 3–5) required to protonate the carboxylate in solution. In proteins, its pK varies from a low of about 2, to a high of nearly 9 (Gunner et al. 2000; Georgescu et al. 2002; Laurents et al. 2003; Li et al. 2004). The higher pK values occur when the protonated carboxyl group is stabilized in a nonpolar environment (Mehler et al. 2002). The protonated carboxyl group has a relatively low ab initio dipole moment of 1.52 D (Table 1). When ionized, the negative charge is likely repelled by the π electron cloud of Chl a. As the LHC apoprotein folds, the approach of a positively charged guanidinium group attracts electron density from the carboxylate group and the ion-pair is stabilized by the nonpolar environment. The resulting glutamate in this ion-pair is a soft Lewis base, with a dipole moment probably near that of a protonated carboxyl group.
The dipole moment of the ion-pair is possibly too low to displace a water molecule from Chl b. With its electron cloud pulled away from the central Mg (∼+1 charge), Chl b is expected to coordinate more readily with a carboxyl group (∼−1 charge) and thus form a bond with largely electrostatic character. In LHCII, Chl b is coordinated with the sidechain of Glu139 near the stromal end of helix-2, which nevertheless is sufficiently near Arg142 for at least partial charge-compensation (Standfuss et al. 2005).
The ability of Chls to bind to imidazole and glutamate/arginine ion-pair ligands was assayed experimentally by interaction with a synthetic peptide that mimicked helix-1 in LHCPs. Chls a and d bound with nearly equal affinity, assayed by Förster resonance energy transfer from a tryptophan residue next to arginine (Chen et al. 2005). In contrast, Chls b and c did not interact significantly with the peptide. A theoretical analysis of the interaction of the Chls with the peptide (Chen and Cai 2007) strongly supports the experimental data and indicates that bonding of Chls b and c with these ligands is thermodynamically unfavorable (Table 2). In the presence of water, however, complex formation with Chl b is more favorable, which, as shown by molecular modeling, is the result of a water molecule bridging Chl b and the ligand (Chen and Cai 2007).
Amide group
The pK of an amide is −0.62 (Grant et al. 1983), an indication that electrons on the oxygen or nitrogen are not readily available for bonding with H+. However, the group exhibits a relatively strong dipole, with the negative end on the oxygen atom. Sidechain amides in proteins have a dipole moment of 3.46 D (Table 1). The dipole moments of the model compounds, formamide and acetamide, are about 3.8 D in solution. The dipole is sufficiently strong to displace a coordinated water molecule from Chl a and should also allow effective competition with water for Chl b within the environment of a membrane. In reconstituted complexes, several sites, including Gln197, have mixed occupancy (Bassi et al. 1999; Remelli et al. 1999), which suggests competition during folding. Yet the sidechain amide groups of Asn183 and Gln197 are ligands for only Chl a in LHCII in vivo, which suggests that Chl a is more abundant during folding and competes effectively by mass action.
Peptide bond
Backbone amides are usually H-bonded within helical or β-sheet structures in proteins and therefore unavailable for interaction with Chls. However, proline residues occur at conserved regions within Chl-binding proteins, particularly in the N-terminal region of LHCPs (Jansson 1999), which preclude formation of H-bonds to nearby backbone carbonyl groups. The pK value for the model compound N-methylacetamide, an analog of the peptide bond, is −0.42 (Grant et al. 1983), slightly more basic than a sidechain amide group. The electron-donating methyl group bonded to the nitrogen provides a larger “pool” of electrons for the carbonyl oxygen to draw from, which is reflected in the slightly higher pK value than of the sidechain amide group. This effect also increases the dipole moment of the carbonyl group. N-Methylacetamide has a calculated dipole of 3.73 D in the gas phase and a monomeric dipole moment of 4.2 D in liquid, which is the same as the calculated dipole moment of the peptide bond in proteins (Table 1). The ab initio dipole moment of the fully H-bonded N-methylacetamide in liquid is calculated to have an average of 6 D, with a spread from 4 to 8 D (Whitfield et al. 2006). Urea has an even stronger dipole moment than N-methylacetamide and also illustrates the effect of H-bonding. Its molecular dipole moment is 5.15 D and is increased to 7.04 D in the crystal form (Abramov et al. 1999). An electrophilic center such as the Mg atom of Chl should have a similar effect to that of H-bonding. As expected from the strong dipole moment of urea, the negative point charge on its carbonyl oxygen may be repulsed by the electron density enclosing the Mg of Chl a, which prevents it from being an effective ligand. Urea does not seem to compete with imidazole or the glutamate/arginine ion-pair, the most favorable ligands of Chl a (Eggink and Hoober 2000). However, it should effectively compete with other ligands for binding to Chl b.
A peptide bond carbonyl in association with a polarizing, positively charged Mg should have a dipole moment at least as large as H-bonded N-methylacetamide and therefore should form a strong electrostatic bond with Chl b. A free backbone carbonyl group in helix-1 in Lhcb1 occurs at Gly78, because of nearby Pro82, within the interior of the membrane. This group should displace water from a Chl molecule but is probably sterically hindered from coordinating directly with the Mg atom by the adjacent, bulky amino acid sidechains of leucine and phenylalanine (see Fig. 3). As a result, this ligand is bridged by a water molecule to Chl a6 in LHCII (Standfuss et al. 2005). That this position is occupied by Chl a rather than Chl b may be determined by the availability and order of binding of the Chls during assembly of the complex. Repulsion of the strong dipole by the electron cloud of Chl a possibly limits its direct interaction with backbone carbonyl groups.
Of particular interest is the finding that backbone carbonyls of proline residues provide ligands to Chl a in a water-soluble Chl-binding protein from Lepidium virginicum (Horigome et al. 2007). In the complex, four Chl a molecules are bound in a solvent-excluded pocket at the interfaces of the tetrameric protein. This observation is evidence that Chl a can fulfill the full range of ligand coordination with sufficient support from the local environment. However, this arrangement is quite different from the interaction of monomeric Chls with LHCPs during LHC assembly.
Phosphatidyl glycerol
An oxygen of the phosphodiester linkage in a phosphatidyl glycerol molecule serves as the ligand in LHCII to Chl a7, which resides near the stromal surface of the thylakoid membrane (Liu et al. 2004). This rare ligand is also found in photosystem (PS) I (Jordan et al. 2001). The side of the phosphodiester group opposite the Chl a molecule is H-bonded to sidechains of tyrosine and lysine residues (Liu et al. 2004), which probably reduces the electron density on the ligand oxygen. Approach of the negative end of a strong dipole toward the Mg in Chl a should again be hindered by the electron density surrounding the metal.
Implications for assembly of light-harvesting complexes
The concept of ligand preference was developed largely because Chl b is found only in LHCs, at specific sites, and with rare exceptions is not found in core complexes. X-ray diffraction studies of crystallized reaction centers (Fromme et al. 2001; Jordan et al. 2001; Loll et al. 2005) and LHCII (Liu et al. 2004; Standfuss et al. 2005) revealed unambiguous Chl-ligand pairs, without mixed occupancy. Whereas a nitrogen atom of the imidazole group of histidine is the most common ligand, sidechain amide groups, water molecules and even a few carboxylate groups occur as ligands of Chl a in reaction centers of PS I and PS II (Jordan et al. 2001; Balaban 2005; Balaban et al. 2002; Oba and Tamiaki 2002, 2005; Ferreira et al. 2004).
Although Chl b expresses more strongly a positive charge on the central Mg than Chl a, and consequently interacts electrostatically more readily with hard Lewis bases, the above analysis indicates that Chl a can also bind to these ligands. However, a stable LHCII cannot be reconstituted in vitro with only Chl a. Conversely, stable LHCII was reconstituted with only Chl b, and the number of Chl b molecules (13.5) was the same as when both Chls were present (13.7) (Kleima et al. 1999; Reinsberg et al. 2001; Schmid et al. 2001). Thus sites normally occupied by Chl a can be occupied by Chl b. Whether Chl b molecules indeed interact directly with ligands of Chl a or are bridged by water molecules remains to be determined. As shown in Table 2, calculated ΔH of formation of complexes with a synthetic peptide containing the glutamate/arginine ion-pair and a histidine residue indicated that complex formation with Chl b is thermodynamically unfavorable. However, insertion of a water molecule to bridge Chl b and the ligand dramatically increased thermodynamic stability (Chen and Cai 2007).
The overlap of ligand characteristics is substantial and too much may have been made about ligand selectivity with Chls. Therefore, unambiguous occupancy of specific binding sites as found in vivo must involve more than these properties. Two recent publications shed light on how the LHCII complex is assembled in vivo. Reinbothe et al. (2006) showed that LHCPs are not imported at a detectable rate into plastids purified from a Chl b-less mutant of Arabidopsis thaliana. These authors also confirmed the localization of CAO on the inner membrane of the chloroplast envelope as reported by Eggink et al. (2004). An alternate site for CAO activity was achieved by Hirashima et al. (2006), who transformed the Chl b-less mutant of A. thaliana ch1-1 with the gene for CAO from the cyanobacterium Prochlorothrix hollandica to achieve active CAO on thylakoid membranes. In these plants, the higher Chl b content resulted in a Chl a/b ratio that approached 1, much lower than the ratio of 3–4 in wild-type plants. In LHCII, the ratio was 0.8 in contrast to 1.3 in the complex from wild-type plants. Chl b was recovered in purified PSI and PSII core complexes, which normally lack Chl b, as well as in LHCs. Because of the widespread distribution of Chl b in the transformed plants, Hirashima et al. (2006) concluded that the restrictive distribution of the Chls in wild-type plants is not the result of discriminatory binding affinities of Chl a and Chl b to ligands. Because the P. hollandica CAO on thylakoid membranes led to a widespread distribution of Chl b, the conclusion emerges that active CAO only on the envelope of chloroplasts leads to restriction of Chl b to LHCs. To achieve incorporation of Chl b selectively into LHCs, as found in wild-type plants, assembly of LHCs should therefore occur during import at the level of the envelope.
In Chl b-less mutants of higher plants, only a few of the apoproteins for LHCI and LHCII accumulate in the organelle in vivo (Król et al. 1995; Bossmann et al. 1997; Espineda et al. 1999). This observation has traditionally been interpreted as an indication that the proteins are rapidly degraded upon entry into the chloroplast stroma unless Chl b is present to allow stable integration into the thylakoid membrane. However, as noted above, chloroplasts from a Chl b-less mutant of A. thaliana lacked the ability to import LHCPs. Accumulation of LHCPs in vivo into the plastid of the alga Chlamydomonas reinhardtii was markedly reduced in the Chl b-less strain, cbn1-113 (Park and Hoober, 1997). Mature-sized proteins were detected in the cytosol, which indicated that import was aborted (White et al. 1996; Park and Hoober 1997). In the absence of Chl, a condition achieved because the mutant strain was unable to synthesize Chl in the dark, accumulation of LHCPs in the plastid was not detected. However, the proteins were synthesized at the same rate as in cells greening in the light but accumulated in the cytosol and vacuoles. These results point to a requirement of Chl, and particularly of Chl b, for import and/or retention of LHCPs in the organelle. Even in wild-type cells, excess LHCPs were shunted to vacuoles when the rate of Chl synthesis was insufficient to accommodate the rate of synthesis of the Chl-binding proteins (White et al. 1996).
Model of LHCII assembly
Folding of a thylakoid membrane protein of cytosolic origin is a complex process, made more so by the environmental sensing of domains as the protein is threaded through the translocon in the chloroplast envelope. Popot and Engelman (2000) and Bowie (2005) described two steps of the process of folding of a membrane protein. The first involves achieving the correct location and topology by the initially inserted segments. Second is the folding and condensation of the protein from this starting point. The evidence indicates that LHCP precursors achieve location and topology as they are guided by the transit sequence through translocons on the outer and inner envelope membranes. The transit sequence is removed soon after the N-terminal domain gains access to the stroma (Soll and Schleiff 2004; Vothknecht and Soll 2006). Membrane-spanning, nonpolar sequences that serve as stop-transfer domains are minimally 14–16 amino acids in length (Davis and Model 1985; Adams and Rose 1985; Popot and Engelman 2000). In this respect, assembly of LHCs presents an interesting problem. Inspection of helix-1 of most LHCPs reveals that the length of the nonpolar sequence in the first membrane-spanning domain is only 10–12 amino acids long, which is on the short side of a significant stop-transfer signal. The nonpolar sequence is within the lagging half of the span, terminated by several charged amino acids (Green and Durnford 1996; Jansson 1999). Binding of Chl to amino acids in the leading half of the helix, which is untypically polar and charged for a membrane-spanning segment of a protein (see Fig. 3), should increase the probability that this domain remains in the membrane.
It seems plausible to consider that as the N-terminal domain of LHCPs traverses the envelope inner membrane, sidechains of glutamate and arginine within the conserved sequence –EVIHSR– in helix-1 form a looped ion-pair ligand for Chl a. The histidine residue provides a second ligand for Chl a, as described by Eggink and Hoober (2000). Kohorn (1990) showed that mutation of this sequence, to replace histidine with alanine, eliminated the ability of the chloroplast to import a LHCP precursor. Binding of Chl to these sidechains may allow this polar sequence to diffuse more readily into the nonpolar phase of the membrane. However, these interactions are insufficient to retain the protein in the membrane in the absence of Chl b. As shown by Chen et al. (2005), Chls b and c bind poorly to imidazole or glutamate/arginine ion pairs in vitro.
A possible ligand for the Chl b molecule that is necessary for retention of the protein in the plastid was suggested by the crystal structure of LHCII. Several backbone carbonyls near the N-terminus are precluded from H-bonding and formation of an α-helix because of the richness of proline residues in this region of the LHCP. The carbonyl of tyrosine (Tyr24 in spinach Lhcb1) resides three positions distant in the amino acid sequence from a proline residue and is thus free to form a coordination bond with Chl b (Liu et al. 2004; Standfuss et al. 2005). The unusual abundance of proline in the N-terminal domain also extends to an iron-deficiency-induced (Tidi) protein, a homolog of the light-harvesting Chl a/b proteins, in Dunaliella, which increases the probability of interaction with Chl b during this stress condition that leads to chlorosis (Varsano et al. 2006). The strong electrostatic bond formed by further polarization of the carbonyl dipole through interaction with Chl b may be essential to anchor a LHCP in the envelope membrane sufficiently long for the remainder of the protein to be transported from the cytosol to complete assembly (Fig. 4).

Model of LHCII assembly in the chloroplast envelope and the proposed role of Chl b. Several proposed intermediates are shown in the sequence, left to right. After synthesis in the cytosol, a LHCP precursor is imported sufficiently into the chloroplast stroma for removal of the transit sequence from the N-terminus and for the first membrane-spanning region to engage the inner membrane. Chl a (dark green rectangles) binds to ligands in the motif provided by the ion-pair of the sidechains of glutamate and arginine and the imidazole group of histidine (dotted line, a). However, binding to these sites is not sufficient to retain the protein in the envelope. Without Chl b the protein slips back into the cytosol for transfer to vacuoles and subsequent degradation. Chl b (light green rectangles) forms a strong coordination bond with the peptide bond carbonyl of Tyr24, near the N-terminus, and provides an additional hold on the protein (solid line, a + b). Along with the Chl a molecules that bind to the motifs in membrane-spanning helix 1, Chl b binds to Try24 and the peptide carbonyl of Val119 at the lumenal end of helix-2. These Chls retain the protein in the membrane sufficiently long for the remainder of the protein, including the conserved motif in membrane-spanning helix-3, to enter the membrane, bind additional Chl and xanthophylls molecules, and complete assembly (LHC). Other proteins in the membrane and stroma apparently assist assembly of the complete complex (see text)
As the remainder of LHCP is transferred through the translocon, a second Chl b possibly coordinates with the backbone carbonyl of Val119 (Liu et al. 2004; Standfuss et al. 2005). Helices 2 and 3 are then transferred through the membrane, which would complete the first step described above by Bowie (2005). The rather weak hydrophobic character of helix-2, and the short nonpolar sequence (again the lagging half) in helix-3, suggest that these domains may enter the membrane largely unassisted, as found for other membrane proteins containing transmembrane sequences that are only moderately hydrophobic (Brambillasca et al. 2006). Other factors are also required at this step, probably to prevent helix-3 from escaping the membrane into the stroma (Fig. 4). One of these factors is the chloroplast signal-recognition particle. The ability of this complex to bind with high affinity to the loop between helices 2 and 3 (Tu et al. 2000) suggests that it plays an important role in this process (Schünemann 2003). Other proteins such as Albino3 have been identified as important in the integration of LHCPs into the membrane (Moore et al. 2000; Bellafiore et al. 2002), although their specific actions are not known. Gerdes et al. (2006) found that Alb3 mutants were defective in chloroplast biogenesis but not in accumulation of LHCPs. With the overall disposition of the protein now achieved in the membrane, the glutamate/arginine ion-pairs between helices 1 and 3 can now form, which stabilizes the protein.
Although the pK values vary widely, the magnitude of the dipole moments of the ligands that selectively coordinate with Chl a or Chl b are not substantially different, except for the backbone carbonyl group. Therefore, as Hirashima et al. (2006) conclude, highly specific ligand selectivity should not be expected. In vitro reconstitution of LHCs has been remarkably successful in reflecting the innate stability of Chl-ligand pairs (Bassi et al. 1999; Remelli et al. 1999; Rogl and Kühlbrandt 1999; Horn and Paulsen 2004) but these experiments did not fully achieve the selectivity of interaction that is found in complexes that are assembled in vivo. Ligand selection can reasonably be considered by taking into account (i) the unique Lewis acid properties of Chl b, (ii) the preference of Chl b to form electrostatic bonds with hard ligands containing a fixed dipole (i.e., an oxygen atom), (iii) the micro-environments in which the interactions occur (i.e., the dielectric constant), and (iv) the order of addition of the pigment molecules, which is determined partly by the local concentrations of the two Chls. Interaction of Chl b with sidechain amide groups would be expected from the properties of the ligands, and both Chl a and b are found with these ligands after in vitro reconstitution of LHCs (Remelli et al. 1999). Yet these groups are not ligands of Chl b in the crystal structure of LHCII. Chl a is probably more abundant within the membrane and competes favorably for amide ligands by mass action during assembly. Thus, basing theoretical proposals for a specific Chl in each binding site on only the first three factors above is not sufficient. The lack of ambiguity, or mixed sites, found in complexes isolated from plants after in vivo assembly is most likely achieved also by the order in which Chls bind, determined by the relative concentrations of each.
An important aspect in the interaction of Chls and ligands is the location of the ligand within the protein structure. Tyr24 is near the stromal surface of LHCII, in a region expected to have a membrane interface dielectric constant of 5–10 (Tanizaki and Feig 2005). As suggested below, association of Chl b9 with this residue may form prior to reaching this position in the membrane. Likewise, the carbonyl of Val119, non-H-bonded because of Pro116, is at the lumenal end of helix-2 in LHCII, also a region of relatively high dielectric constant. The backbone carbonyl group of Val119 coordinates with Chl b14. The Chl b molecules are possibly protected from the aqueous environment by the protein strand. H-Bonding of the 7-formyl group to Gln122 and Ser123 (Liu et al., 2004) would increase the Lewis acid strength of Chl b14 and strengthen this bond. These two Chl b molecules possibly stabilize an intermediate in the assembly pathway of LHCII (Fig. 4).
Four Chl b molecules interact with helix-2 of Lhcb1. Chl b12 forms a coordination bond with Glu139, which as discussed above is likely charge-compensated by Arg142. The other three Chl b molecules (b10, b11 and b13) retain water ligands (Fig. 3). H-Bonding of these Chl b molecules through the 7-formyl group would further enhance the electrostatic character of the Mg atom (Liu et al. 2004). The 7-formyl group of Chl b11 is H-bonded to the peptide bond N of Leu148, and along with Chl b12 may help to retain the stromal end of helix-2 in the membrane. The 7-formyl group of Chl b13 is H-bonded to the water ligand of Chl b10, which is in turn H-bonded via its 7-formyl group to the amide N of Gln131. These Chl b molecules, as an aggregate, may fill a void in the protein between helices 2 and 3 as the protein folds. Such an aggregate of Chl b molecules would be considerably more stable than a similar complex composed of Chl a. These molecules would then enter the structure late in assembly, as Horn and Paulsen (2004) and Horn et al. (2007) found during studies of the kinetics of reconstitution of the complex in vitro.
The argument is then reduced to one or two critical Chl b molecules that are required for retention of LHCPs in the chloroplast envelop during assembly–one that interacts with a backbone carbonyl near the N-terminus and possibly the second that binds to the lumenal end of helix-2. Since the catalytic center of CAO is on the envelope inner membrane facing the intermembrane space (Reinbothe et al. 2006), the N-terminal domain of LHCP possibly binds to Chl b on that side of the membrane. As the protein is transported through the membrane, the bound Chl b would approach the stromal surface while the motif –ExxHxR– in helix-1 enters the interior of the membrane and binds Chl a (Fig. 4). These Chl molecules may then hold the N-terminal domain in the inner membrane sufficiently long for the remainder of the protein to be transported across the outer membrane of the envelope and become integrated into the inner membrane. This scenario ensures that Chl b enters the complex from the surface of the inner membrane that faces the outer membrane and thus occurs only in peripheral LHCs that are assembled with apoproteins synthesized in the cytosol.
Abbreviations
- BChl:
-
Bacteriochlorophyll
- CAO:
-
Chlorophyllide a oxygenase
- Chl:
-
Chlorophyll
- Chlide:
-
Chlorophyllide
- D:
-
Debye
- LHC:
-
Light-harvesting complex
- LHCP:
-
Light-harvesting complex apoproteins
- MCD:
-
Magnetic circular dichroism
- Pchlide:
-
Protochlorophyllide
- PS:
-
Photosystem
References
Abramov YuA, Volkov AV, Coppens P (1999) On the evaluation of molecule dipole moments from multipole refinement of X-ray diffraction data. Chem Phys Lett 311:81–86
Adams GA, Rose JK (1985) Structural requirements of a membrane-spanning domain for protein anchoring and cell surface transport. Cell 41:1007–1015
Akiyama M, Miyashita H, Kise H, Watanabe T, Mimuro M, Miyachi S, Kobayashi M (2002) Quest for minor but key chlorophyll molecules in photosynthetic reaction centers—unusual pigment composition in the reaction centers of the chlorophyll d-dominated cyanobacterium Acaryochloris marina. Photosynth Res 74:97–107
Antoine R, Compagnon I, Rayane D, Broyer M, Dugourd P, Breaux G, Hagemeister FC, Pippen D, Hudgins RR, Jarrold MF (2002) Electric susceptibility of unsolvated glycine-based peptides. J Am Chem Soc 124:6737–6741
Bachvaroff TR, Puerta MVS, Delwiche CF (2005) Chlorophyll c-containing plastid relationships based on analyses of a multigene data set with all four chromalveolate lineages. Mol Biol Evol 22:1772–1782
Balaban TS (2005) Relevance of the diastereotopic ligation of magnesium atoms of chlorophylls in the major light-harvesting complex II (LHC II) of green plants. Photosynth Res 86:251–262
Balaban TS, Fromme P, Holzwarth AR, Krauss N, Prokhorenko VI (2002) Relevance of the diasterotopic ligation of magnesium atoms of chlorophylls in photosystem I. Biochim Biophys Acta 1556:197–207
Ballschmitter K, Cotton TM, Katz JJ (1969) Chlorophyll–water interactions. Hydration, dehydration and hydrates of chlorophyll. Biochim Biophys Acta 180:347–359
Bassi R, Croce R, Cugini D, Sandonà D (1999) Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc Natl Acad Sci USA 96:10056–10061
Batista ER, Xantheas SS, Jónsson H (1998) Molecular multipole moments of water molecules in ice I h . J Chem Phys 109:4546–4551
Bellafiore S, Ferris P, Naver H, Göhre V, Rochaix JD (2002) Loss of Albino3 leads to the specific depletion of the light-harvesting system. Plant Cell 14:2303–2314
Bhattacharya D, Medlin LK (2004) Dating algal origin using molecular clock methods. Protist 155:9–10
Bossmann B, Knoetzel J, Jansson S (1997) Screening of chlorina mutants of barley (Hordeum vulgare L.) with antibodies against light-harvesting proteins of PSI and PSII: absence of specific antenna proteins. Photosynth Res 52:127–136
Bowie JU (2005) Solving the membrane protein folding problem. Nature 438:581–589
Brambillasca S, Yabal M, Makarow M, Borgese N (2006) Unassisted translocation of large polypeptide domains across phospholipids bilayers. J Cell Biol 175:767–777
Cai ZL, Crossley MJ, Reimers JR, Kobayashi R, Amos RD (2006) Density functional theory for charge transfer: the nature of N-bands of porphyrins and chlorophylls revealed through CAM-B3LYP, CASPT2, and SAC-CI calculations. J Phys Chem B 110:15624–15632
Chen M, Cai ZL (2007) Theoretical study on the thermodynamic properties of chlorophyll d-peptides coordinating ligand. Biochim Biophys Acta, in press
Chen M, Eggink LL, Hoober JK, Larkin AWD (2005) Influence of structure on binding of chlorophylls to peptide ligands. J Am Chem Soc 127:2052–2053
Croce R, Canino G, Ros F, Bassi R (2002) Chromophore organization in the higher-plant photosystem II antenna protein CP26. Biochemistry 41:7334–7343
Davis NG, Model P (1985) An artificial anchor domain: hydrophobicity suffices to stop transfer. Cell 41:607–614
De Martino A, Douady D, Quinet-Szely M, Rousseau B, Crépineau F, Apt K, Caron L (2000) The light-harvesting antenna of brown algae. Highly homologous proteins encoded by a multigene family. Eur J Biochem 267:5540–5549
Desyatnyk O, Pszczólkowski L, Thorwirth S, Krygowski TM, Kisiel Z (2005) www.rsc.org/pccp, accessed March 26, 2006
Dudev T, Cowan JA, Lim C (1999) Competitive binding in magnesium coordination chemistry: water versus ligands of biological interest. J Am Chem Soc 121:7665–7673
Durnford DG, Deane JA, Tan S, McFadden GI, Gantt E, Green BR (1999) A plylogenetic assessment of the eukaryotic light-harvesting proteins, with implications for plastid evolution. J Mol Evol 48:59–68
Dyke TR, Muenter JS (1973) Electric dipole moments of low J states of H2O and D2O. J Chem Phys 59:3125–3127
Eggink LL, Hoober JK (2000) Chlorophyll binding to peptide maquettes containing a retention motif. J Biol Chem 275:9087–9090
Eggink LL, Park HS, Hoober JK (2001) The role of chlorophyll b in photosynthesis: hypothesis. BMC Plant Biol 1:2
Eggink LL, LoBrutto R, Brune DC, Brusslan J, Yamasato A, Tanaka A, Hoober JK (2004) Synthesis of chlorophyll b: localization of chlorophyllide a oxygenase and discovery of a stable radical in the catalytic subunit. BMC Plant Biol 4:5
Epstein J, Ruble JR, Craven BM (1982) The charge density in imidazole by X-ray diffraction at 103 and 293 K. Acta Crystallogr B38:140–149
Espineda CE, Linford AS, Devine D, Brusslan JA (1999) The AtCAO gene, encoding chlorophyll a oxygenase, is required for chlorophyll b synthesis in Arabidopsis thaliana. Proc Natl Acad Sci USA 96:10507–10511
Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S (2004) Architecture of the photosynthetic oxygen-evolving center. Science 303:1831–1838
Frackowiak D, Bauman D, Manikowski H, Browett WR, Stillman MJ (1987) Circular dichroism and magnetic circular dichroism spectra of chlorophylls a and b in nematic liquid crystals. Biophys Chem 28:101–114
Fragata M, Nordén B, Kurucsev T (1988) Linear dichroism (250–700 nm) of chlorophyll a and pheophytin a oriented in a lamellar phase of glycerylmonooctanoate/H2O. Characterization of electronic transitions. Photochem Photobiol 47:133–143
Fromme P, Jordan P, Krauss N (2001) Structure of photosystem I. Biochim Biophys Acta 1507:5–31
Garrido JL, Otero J, Maestro MA, Zapata M (2000) The main nonpolar chlorophyll c from Emiliania huxleyi (Prymnesiophyceae) is a chlorophyll c 2-monogalactosyldiacylglyceride ester: a mass spectrometry study. J Phycol 36:497–505
Georgescu RE, Alexov EG, Gunner MR (2002) Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys J 83:1731–1748
Gerdes L, Bals T, Klostermann E, Karl M, Phillippar K, Hünken M, Soll J, Schünemann (2006) A second thylakoid membrane-localized Alb3/Oxa1/YidC homologue is involved in proper chloroplast biogenesis in Arabidopsis thaliana. J Biol Chem 281:16632–16642
Goss R, Wilhelm C, Garab G (2000) Organization of the pigment molecules in the chlorophyll a/b/c containing alga Mantoniella squamata (Prasinophyceae) studied by means of absorption, circular and linear dichroism spectroscopy. Biochim Biophys Acta 1457:190–199
Grant HM, Mctigue P, Ward DG (1983) The basicities of aliphatic amides. Aust J Chem 36:2211–2218
Gregory JK, Clary DC, Liu K, Brown MG, Saykally RJ (1997) The water dipole moments in water clusters. Science 275:814–817
Green BR, Durnford DG (1996) The chlorophyll-carotenoid proteins of oxygenic photosynthesis. Annu Rev Plant Physiol Plant Mol Biol 47:685–714
Gunner MR, Saleh MA, Cross E, ud-Doula A, Wise M (2000) Backbone dipoles generate positive potentials in all proteins: origins and implications of the effect. Biophys J 78:1126–1144
Gunner MR, Mao J, Song Y, Kim J (2006) Factors influencing the energetics of electron and proton transfer in proteins. What can be learned from calculations? Biochim Biophys Acta 1757:942–968
Hartwich G, Fiedor L, Simonin I, Cmiel E, Schäfer W, Noy D, Scherz A, Scheer H (1998) Metal-substituted bacteriochlorophylls. 1. Preparation and influence of metal and coordination on spectra. J Am Chem Soc 120:3675–3683
Helfrich M, Bommer B, Oster U, Klement H, Mayer K, Larkum AWD, Rüdiger W (2003) Chlorophylls of the c family: absolute configuration and inhibition of NADPH:protochlorophyllide oxidoreductase. Biochim Biophys Acta 1605:97–103
Herschlag D, Jencks WP (1989) Evidence that metaphosphate monoanion is not an intermediate in solvolysis reactions in aqueous solution. J Am Chem Soc 111:7579–7586
Hirashima M, Satoh S, Tanaka R, Tanaka A (2006) Pigment shuffling in antenna systems achieved by expressing prokaryotic chlorophyllide a oxygenase in Arabidopsis. J Biol Chem 281:15385–15393
Hoff AJ, Amesz J (1991) Visible absorption spectroscopy of chlorophylls. In: Scheer H (ed) Chlorophylls. CRC Press, Boca Raton, FL, pp 723–738
Hoober JK, Eggink LL (1999) Assembly of light-harvesting complex II and biogenesis of thylakoid membranes in chloroplasts. Photosynth Res 61:197–215
Hoober JK, Arygroudi-Akoyunoglou JH (2004) Assembly of light-harvesting complexes of photosystem II and the role of chlorophyll b. In: Papageorgiou G, Govindjee (eds) Chlorophyll a fluorescence: A signature of photosynthesis. Advances in photosynthesis and respiration, vol 19. Springer, Dordrecht, The Netherlands, pp 679–712
Horigome D, Satoh H, Itoh N, Mitsunaga K, Oonishi I, Nakagawa A, Uchida A (2007) Structural mechanism and photoprotective function of water-soluble chlorophyll-binding protein. J Biol Chem 282:6525–6531
Horn R, Paulsen H (2004) Early steps in the assembly of light-harvesting chlorophyll a/b complex–time-resolved fluorescence measurements. J Biol Chem 279:44400–44406
Horn R, Grundmann G, Paulsen H (2007) Consecutive binding of chlorophylls a and b during the assembly in vitro of light-harvesting chlorophyll-a/b protein (LHCIIb). J Mol Biol 366:1045–1054
Jansson S (1999) A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci 4:236–240
Jeffrey SW, Wright SW (1987) A new spectrally distinct component in preparations of chlorophyll c from the micro-alga Emiliania huxleyi (Prymnesiophyceae). Biochim Biophys Acta 894:180–188
Jensen WB (1978) The Lewis acid-base definitions: a status report. Chem Rev 78:1–22
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N (2001) Three-dimensional structure of cyanobacterial photosystem 1 at 2.5 Å resolution. Nature 411:909–917
Kleima FJ, Hobe S, Calkoen F, Urbanus ML, Peterman EJG, van Grondelle R, Paulsen H, van Amerongen H (1999) Decreasing the chlorophyll a/b ratio in reconstituted LHCII: structural and functional consequences. Biochemistry 38:6587–6596
Knox RS, Spring BQ (2003) Dipole strengths in the chlorophylls. Photochem Photobiol 77:497–501
Kohorn BD (1990) Replacement of histidines of light harvesting chlorophyll a/b binding protein II disrupts chlorophyll-protein complex assembly. Plant Physiol 93:339–342
Kramer DM, Sacksteder DA, Cruz JA (1999) How acid is the lumen? Photosynth Res 60:151–163
Król M, Spangfort MD, Huner NPA, Öquist G, Gustafsson P, Jansson S (1995) Chlorophyll a/b-binding proteins, pigment conversions, and early light-induced proteins in a chlorophyll b-less barley mutant. Plant Physiol 107:873–883
Laurents DV, Huyghues-Despointes BMP, Bruix M, Thurlkill RL, Schell D, Newsom S, Grimsley GR, Shaw KL, Treviño S, Rico M, Briggs JM, Antosiewicz JM, Scholtz JM, Pace CN (2003) Charge–charge interactions are key determinants of the pK values of ionizable groups in ribonuclease Sa (pI = 3.5) and a basic variant (pI = 10.2). J Mol Biol 325:1077–1092
Li H, Robertson AD, Jensen JH (2004) The determinants of carboxyl pKa values in turkey ovomucoid third domain. Proteins 55:689–704
Linnanto J, Korppi-Tommola J (2004) Semiempirical PM5 molecular orbital study on chlorophylls and bacteriochlorophylls: comparison of semiempirical, ab initio, and density functional results. J Comput Chem 25:123–137
Linnanto J, Martiskainen J, Lehtovuori V, Ihalainen J, Kananavicius R, Barbato R, Korppi-Tommola J (2006) Excitation energy transfer in the LHC-II trimer: a model based on the new 2.72 Å structure. Photosynth Res 87:267–279
Liu Z, Yan H, Wang K, Kuang T, Zhang J, Gui L, An X, Chang W (2004) Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 428:287–292
Loll B, Kern J, Saenger W, Zouni A, Biesiadka J (2005) Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 438:1040–1044
Mehler EL, Fuxreiter M, Simon I, Garcia-Moreno EB (2002) The role of hydrophobic microenvironments in modulating pK a shifts in proteins. Proteins 48:283–292
Miyashita H, Adachi K, Kurano N, Ikemoto H, Chihara M, Miyachi S (1997) Pigment composition of a novel oxygenic photosynthetic prokaryote containing chlorophyll d as the major chlorophyll. Plant Cell Physiol 38:274–281
Moore M, Harrison MS, Peterson EC, Henry R (2000) Chloroplast Oxa1p homolog Albino3 is required for post-translational integration of the light-harvesting chlorophyll-binding protein into thylakoid membranes. J Biol Chem 275:1529–1532
Nagata N, Tanaka R, Satoh S, Tanaka A (2005) Identification of a vinyl reductase gene for chlorophyll synthesis in Arabidopsis thaliana and implications for the evolution of prochlorococcus species. Plant Cell 17:233–240
Nishigaki A, Ohshima S, Nakayama K, Okada M, Nagashima U (2001) Application of molecular orbital calculations to interpret the chlorophyll spectral forms of pea photosystem II. Photochem Photobiol 73:245–248
Noy D, Yerushalmi R, Brumfeld V, Ashur I, Scheer H, Baldridge KK, Scherz A (2000) Optical absorption and computational studies of [Ni]-bacteriochlorophyll-a. New insight into charge distribution between metal and ligands. J Am Chem Soc 122:3937–3944
Oba T, Tamiaki H (2002) Which side of the π-macrocycle plane of (bacterio)chlorophylls is favored for binding of the fifth ligand? Photosynth Res 74:1–10
Oba T, Tamiaki H (2005) Effects of peripheral substituents on diasterioselectivity of the fifth ligand-binding to chlorophylls, and nomenclature of the asymmetric axial coordination sites. Bioorg Med Chem 13:5733–5739
Palmer JD (2003) The symbiotic birth and spread of plastids: how many times and whodunit? J Phycol 39:4–11
Park H, Hoober JK (1997) Chlorophyll synthesis modulates retention of apoproteins of light-harvesting complex II by the chloroplast in Chlamydomonas reinhardtii. Physiol Plant 101:135–142
Phillips JN (1963) Physico-chemical properties of porphyrins. In: Florkin M, Stotz EH (eds) Comprehensive biochemistry, vol 9. Elsevier, Amsterdam, pp 34–72
Popot J-L, Engelman DM (2000) Helical membrane protein folding, stability, and evolution. Annu Rev Biochem 69:881–922
Porra RJ (1997) Recent progress in porphyrin and chlorophyll biosynthesis. Photochem Photobiol 65:492–516
Reinbothe C, Bartsch S, Eggink LL, Hoober JK, Brusslan J, Andrade-Paz R, Monnet J, Reinbothe S (2006) A role for chlorophyllide a oxygenase in the regulated import and stabilization of light-harvesting chlorophyll a/b proteins. Proc Natl Acad Sci USA 103:4777–4782
Reinsberg D, Ottmann K, Booth PJ, Paulsen H (2001) Effects of chlorophyll a, chlorophyll b, and xanthophylls on the in vitro assembly kinetics of the major light-harvesting chlorophyll a/b complex, LHCIIb. J Mol Biol 308:59–67
Remelli R, Varotto C, Sandonà D, Croce R, Bassi R (1999) Chlorophyll binding to monomeric light-harvesting complex: a mutational analysis of chromophore-binding residues. J Biol Chem 274:33510–33521
Rodriguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Löffelhardt W, Bohnert HJ, Philippe H, Lang BF (2005) Monophyly of primary photosynthetic eukaryotes: green plants, red algae and glaucophytes. Curr Biol 15:1325–1330
Rogl H, Kühlbrandt W (1999) Mutant trimers of light-harvesting complex II exhibit altered pigment content and spectroscopic features. Biochemistry 38:2281–2287
Sacksteder CA, Kanazawa A, Jacoby ME, Kramer DM (2000) The proton to electron stoichiometry of steady-state photosynthesis in living plants: a proton-pumping Q cycle is continuously engaged. Proc Natl Acad Sci USA 97:14283–14288
Schmid VHR, Thomé P, Rühle W, Paulsen H, Kühlbrandt W, Rogl H (2001) Chlorophyll b is involved in long-wavelength spectral properties of light-harvesting complexes LHC I and LHC II. FEBS Lett 499:27–31
Schmitt A, Frank G, James P, Staudenmann W, Zuber H, Wilhelm C (1994) Polypeptide sequence of the chlorophyll a/b/c-binding protein of the prasinophycean alga Mantoniella squamata. Photosynth Res 40:269–277
Schünemann D (2003) Structure and function of the chloroplast signal recognition particle. Curr Genet 44:295–304
Shalchian-Tabrizi K, Skånseng M, Ronquist F, Klaveness D, Bachvaroff TR, Delwiche CF, Botnen A, Tengs, Jakobsen KS (2006) Heterotachy processes in Rhodophyte-derived secondhand plastid genes: implications for addressing the origin and evolution of dinoflagellate plastids. Mol Biol Evol 23:1504–1515
Simonetto R, Crimi M, Sandonà D, Croce R, Cinque G, Breton J, Bassi R (1999) Orientation of chlorophyll transition moments in the higher-plant light-harvesting complex CP29. Biochemistry 38:12974–12983
Smith KM (1975) General features of the structure and chemistry of porphyrin compounds. In: Smith KM (ed) Porphyrins and metalloporphyrins. Elsevier Scientific, Amsterdam, pp 1–28
Smith MB, March J (2001) March’s advanced organic chemistry, 5th edn. John Wiley and Sons, New York
Soll J, Schleiff E (2004) Protein import into chloroplasts. Nat Rev Mol Cell Biol 5:198–208
Spackman MA (1992) Molecular electric moments from X-ray diffraction data. Chem Rev 92:1769–1797
Standfuss J, Terwisscha van Scheltinga AC, Lamborghini M, Kühlbrandt W (2005) Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 Å resolution. EMBO J 24:919–928
Sundholm D (2003) A density-functional-theory study of bacteriochlorophyll b. Phys Chem Chem Phys 5:4265–4271
Tamiaki H, Yagai S, Miyatake T (1998) Synthetic zinc tetrapyrroles complex with pyridine as a single axial ligand. Bioorg Med Chem 6:2171–2178
Tanizaki S, Feig M (2005) A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J Chem Phys 122:124706
Tomitani A, Okada K, Miyashita H, Matthijs HCP, Ohno T, Tanaka A (1999) Chlorophyll b and phycobilins in the common ancestor of cyanobacteria and chloroplasts. Nature 400:159–162
Tu CJ, Peterson EC, Henry R, Hoffman NE (2000) The L18 domain of light-harvesting chlorophyll proteins binds to the chloroplast signal recognition particle 43. J Biol Chem 275:13187–13190
University of Southern Maine, www.usm.maine.edu/∼newton/Chy251_253/Lectures/Solvents/Solvents.html (accessed March 24, 2006)
van Gammeren AJ, Hulsbergen FB, Erkelens C, de Groot HJM (2004) Synthetic analogues of the histidine-chlorophyll complex: a NMR study to mimic structural features of the photosynthetic reaction center and the light-harvesting complex. J Biol Inorg Chem 9:109–117
Varsano T, Wolf SG, Pick U (2006) A chlorophyll a/b-binding protein homolog that is induced by iron deficiency is associated with enlarged photosystem I units in the eukaryotic alga Dunaliella salina. J Biol Chem 281:10305–10315
Vothknecht UC, Soll J (2006) Protein import into chloroplasts: who, when, and how? In: Wise RR, Hoober JK (eds) The structure and function of plastids. Advances in photosynthesis and respiration, vol 23. Springer, Dordrecht, The Netherlands, pp 53–74
White RA, Wolfe GR, Komine Y, Hoober JK (1996) Localization of light-harvesting complex apoproteins in the chloroplast and cytoplasm during greening of Chlamydomonas reinhardtii at 38°C. Photosynth Res 47:267–280
Whitfield TW, Crain J, Martyna GJ (2006) Structural properties of liquid N-methylacetamide via ab initio, path integral, and classical molecular dynamics. J Chem Phys 124:94503–94515
Wise RR, Hoober JK, (eds) (2006) The structure and function of plastids. Advances in photosynthesis and respiration, vol 23. Springer, Dordrecht, The Netherlands
Acknowledgment
MC thanks the Australian Research Council (DP045233 and DP0665169) for financial support. We thank Dr. Zheng-Li Cai for computational calculations and molecular modeling.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Hoober, J.K., Eggink, L.L. & Chen, M. Chlorophylls, ligands and assembly of light-harvesting complexes in chloroplasts. Photosynth Res 94, 387–400 (2007). https://doi.org/10.1007/s11120-007-9181-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-007-9181-1