
Abstract
Background
Many biological questions about N availability and the N cycle require knowledge of the abundance and identity of molecules comprising the pool of organic N. Moreover, basic knowledge of the molecular composition of the soil solution can give rise to new hypotheses via data-driven or inductive reasoning.
Scope
This paper examines the composition of organic N molecules in the soil solution. Our perception of organic N in the soil solution is shaped by analytical approaches, and thus I briefly review approaches for sampling and analysis of the soil solution. I give examples of hypotheses generated by knowledge of the molecular composition of organic N and conclude by suggesting priorities for future research.
Conclusions
Studies of the molecular composition of organic N are very much in their infancy. Amino acids, their oligomers and polymers are consistently large components of the pool of organic N. The soil solution also contains organic N compounds from at least another 12 compound classes, but almost nothing is known about their functional significance. Uncovering the role of these other compounds in the N cycle would enrich our understanding of organic N and the N cycle, and place studies of amino acids and their polymers in a broader context.
Similar content being viewed by others

Introduction
Our view of organic N in the soil solution is constantly evolving (Paungfoo-Lonhienne et al. 2012). For example, one obtains a vastly different picture of organic N molecules in the soil solution in comparing the older literature with much of the past 20 years of research. Experiments from the late 19th century until the 1980s reported occurrence in soil of many different forms of organic N (e.g. protein amino acids, non-protein amino acids, nucleic acids, ureides, quaternary ammonium compounds, purine and pyrimidine bases)(Schreiner 1912; Schreiner and Skinner 1912; Lathrop 1917; Dadd et al. 1953; Aseeva et al. 1977; Aseeva et al. 1978; Schlimme and Kirse 1983) and at least some of these were known 100 years ago to be taken up by plants (e.g. see review by: Hutchinson and Miller 1912). In contrast, for the last 20 years research has focussed on bulk properties of organic N in soil solution (e.g. measurements of DON, C:N ratios), or characterising in detail specific fractions of organic N namely protein amino acids (Chapin et al. 1993; Jones and Darrah 1993; Näsholm et al. 1998; Schimel and Bennett 2004; Warren 2006) and their oligomers and polymers (Friedel and Scheller 2002; Yu et al. 2002; Farrell et al. 2011b; Hill et al. 2011; Hill et al. 2012).
Describing bulk properties of organic N and characterising in detail specific fractions has provided profound insights into the functioning of organic N in soils (e.g. see reviews: Murphy et al. 2000; Chen and Xu 2008), but our understanding is limited in scope and often lacking in context. For example, studies characterising functions of amino acids are difficult to place in the broader context of organic N and the N cycle because amino acids are not the only class of organic N in soil solution (Schreiner 1912; Schreiner and Skinner 1912; Lathrop 1917; Dadd et al. 1953; Aseeva et al. 1977; Aseeva et al. 1978; Schlimme and Kirse 1983). To paraphrase a recent review on dissolved organic matter in marine systems (Mopper et al. 2007): understanding the function of dissolved organic N in soils has been hindered by methodologies in which only bulk parameters were quantified (e.g. pool of DON) or for which a significant pool of organic N resided outside our analytical window (e.g. studies that focus solely on amino acids and their polymers), with both approaches providing incomplete, incorrect or biased insights.
This review begins by establishing some of the reasons that organic N in soils matters, addresses the germane question of what can be learnt by studying pools (as opposed to fluxes) before reviewing the current state of knowledge of organic N molecules in the soil solution. Our perception of what is in the soil has been shaped by the approaches used for sampling and analysis of samples, and thus I briefly review approaches for sampling and analysis of the soil solution. Examples are given where knowledge of the molecular composition of organic N can be used to generate hypotheses about the N cycle. Finally, I conclude by suggesting priorities for future research. The role of organic N in plant nutrition and the N cycle are addressed, but only in the context of hypotheses arising from knowledge of pools of organic N. Readers are referred to other papers for a broader review of the N cycle (Galloway et al. 2008; Gruber and Galloway 2008; Rennenberg et al. 2009), and organic N uptake by plants (Näsholm et al. 2009; Paungfoo-Lonhienne et al. 2012).
Why does organic N in soils matter?
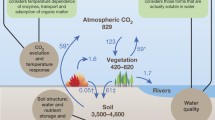
Management of soil nitrogen is one of the keys to sustainably supplying the world with agricultural and forest products. The agricultural and forest industries are under increasing pressure to produce more products from less land. The world’s population is projected to increase by 2.5 billion over the next 43 years, from 6.7 billion to 9.2 billion (Anonymous 2006). The production of agricultural and forest products needs to keep pace not only with the rising population, but also with the increasing standards of living that are accelerating demand for plant products. One of the keys to meeting the challenge is management of nutrients, in particular nitrogen. Nitrogen is one of the nutrients most commonly managed because it is required in large amounts and commonly limits growth. Productivity is increased by applying nitrogenous fertiliser, but this causes significant environmental problems and is costly. For example, globally 85–90 million tons of nitrogenous fertilisers are applied every year to soil (Vitousek et al. 1997; Tilman 1999; Galloway et al. 2008; Gruber and Galloway 2008), which leads to remarkable increases in productivity of forests and crops (e.g. Judd et al. 1996; Smil 2000). The significant economic and environmental costs associated with fertiliser use are a strong incentive to optimise its application by learning how to strike a scientifically informed balance between too little and too much fertiliser. One of the keys to understanding N availability is low molecular weight (MW) organic molecules of the soil solution. This is because low MW organic molecules can be directly taken up by plants (Hutchinson and Miller 1912; Virtanen and Linkola 1946; Paungfoo-Lonhienne et al. 2012) and are intermediates in the breakdown of polymers to release inorganic nutrients (Fig. 1).

Central role of low molecular weight (MW) organic compounds in plant nutrition and biogeochemical cycling
An additional practical reason for studying organic N in the soil solution is their role in the greenhouse gas budget of Earth (Gardenas et al. 2011; Knicker 2011). Soils are major players in the global C budget with a global standing stock of 1500 Pg C and 60 Pg C yr−1 exchange of C between soil and atmosphere (~ ten times larger than flux of C due to burning of fossil fuels) (Amundson 2001; Filley and Boutton 2006). Much of the enormous exchange of C between soils and atmosphere is due to cycling of low MW organic compounds in the soil solution (Schimel and Bennett 2004). For example, it was estimated that 30 % of the total CO2 respiration from soil can be attributed to turnover of low MW organic compounds (van Hees et al. 2005), many of which contain N (Knicker 2011). In addition to DON molecules having a C backbone that directly links their metabolism to the C cycle, there are a suite of less direct ways that DON can influence the C cycle (Gardenas et al. 2011; Knicker 2011).
Fluxes of N are important, so what can be learnt by studying pools?
The biological importance of different organic N compounds cannot be deduced solely from their concentrations in soil solution. For example, small pools of organic N monomers and oligopeptides could be interpreted as evidence that they are “unimportant” for the N cycle, but small concentrations could alternatively be seen as evidence that flux through those forms of N is rapid because they are preferred sources of N for microbes and/or plants. Similarly, small concentrations of amino sugars in the soil solution (Warren 2013c) are consistent with very rapid cycling and removal from the soil solution by high-affinity transporters in microbes (Roberts et al. 2007; Roberts and Jones 2012). In light of the challenges of interpreting what is meant by soil solution concentrations, is it worthwhile reporting or discussing concentrations of different organic N compounds?
Knowledge of identity and concentration of organic N molecules is a pre-requisite for designing and interpreting experiments to quantify fluxes. Irrespective of whether the quantified flux is uptake by plants or cycling by soil microbes the common approaches for measuring fluxes of N involve addition of a certain amount of substrate (typically isotope labelled). Thus choices need to be made as to the identity and concentration of the substrate, and it would make sense if this were informed by what is in the soil solution. In many cases there will be practical reasons why it is not possible to supply substrates at the same concentration as in the soil solution (e.g. larger concentrations are required to ensure detection), but this does not negate the imperative to know what is in the soil solution. Instead the utility of knowing what is in soil solution shifts from designing realistic experiments, to its necessity for interpreting data from sometimes unrealistic experiments. For example, preliminary findings from a recent study of peptides in soil (Warren 2013a) indicate that studies investigating plant uptake of peptides (Hill et al. 2011; Soper et al. 2011) used individual peptides at concentrations that were three (Hill et al. 2011) or even six (Soper et al. 2011) orders of magnitude greater than for the same peptides in soil. Clearly, the debate about organic nutrition of plants cannot move forward until the design and interpretation of experiments is informed by knowledge of the organic N molecules in the soil solution.
In the most general sense, one imperative for knowing the different molecular forms of organic N in the soil solution is that observational data can be used to generate hypotheses via inductive (or data-driven) reasoning (Kell and Oliver 2004). In this sense hypotheses are the outcome of observational data rather than its starting point. To date attempts to understand the origin, function and fate of organic N molecules have adopted reductionist hypothesis-driven means. For example, targeted measurements of concentrations and/or fluxes of selected amino acids. The logic being that one can reconstruct function of the overall system by dividing the system into its component parts (e.g. molecules or classes of molecules) and studying their function. Studies of amino acid fluxes or turnover, for example, do not capture all of the compounds amino acids are known to directly interact with (e.g. amines and fatty acids via putrefaction reactions and peptides by condensation and hydrolysis) or more complex interactions (e.g. as substrates in purine synthesis, pyrimidine interconversions). Hence, reductionist experiments on individual compound classes are unlikely to reveal the complex interactions among compounds and compound classes. In contrast, examining the diversity of organic N molecules and studying the system as a whole is more likely to reveal the complexities and interactions of organic N. Such data can provide a fertile basis for generation of hypotheses that can be tested with subsequent reductionist studies. For example, one can generate hypotheses that make sense of the observations by mapping molecules onto metabolic maps, or organising molecules based on origin (plant, fungal, bacterial) or putative function (osmolytes, primary metabolites, etc.).
What do we know about organic N in soil? Molecular weights and compound classes
For at least some of the major classes of DON we have a reasonable idea of total pool size (Table 1), although limitations of techniques mean that boundaries between quantified classes are sometimes blurred (see discussion below). Soluble N in the soil solution or soil extracts can be broadly separated into organic (DON, dissolved organic N) and inorganic pools (DIN, dissolved inorganic N), with many studies reporting that the pool of DON was of a similar size or larger than DIN (Jones and Kielland 2002; Yu et al. 2002; Jones et al. 2005; Chen and Xu 2008). Studies comparing sites varying in fertility generally indicate that infertile sites have large amounts of DON and small amounts of DIN while fertile sites have large amounts of DON and large amounts of DIN (Nordin et al. 2001; Yu et al. 2003; Christou et al. 2005; Kranabetter et al. 2007). But what do we know about the molecules that comprise DON?
Acid hydrolysis followed by colorimetric and fluorometric assays indicate hydrolysable amino acids (i.e. peptides and proteins) commonly account for at least 50 % of DON, whereas (free) primary amino acids are typically less than 5 % of the pool of DON (Stevenson 1982; Yu et al. 2002; Paul and Williams 2005; Christou et al. 2006)(Fig. 2). The dominance of the soil solution by oligomers and polymers of amino acids is supported by studies with 15N NMR (nuclear magnetic resonance) reporting that peptide-like and protein-like signals dominate DON (Michalzik and Matzner 1999; Kögel-Knabner 2006). In fire-affected ecosystems, 13C and 15N NMR indicates occurrence of a variety of heterocyclic structures (Knicker et al. 2005; Knicker 2007; Knicker 2011), but the low sensitivity of NMR and low N concentrations in soils makes it difficult to perform a quantitative comparisons of proteins and peptides versus heterocyclic structures.

The approximate distribution of dissolved organic N (DON) among broad size classes (left) and the main classes of monomeric DON (right). The broad size-class distribution of DON was approximated from published data for several grassland soils (Farrell et al. 2011a) and is consistent with observations that a larger proportion of DOC is in the <1 kDa fraction than the 1–10 kDa fraction (Marschner and Kalbitz 2003) and most DON is oligomers and polymers of amino acids (e.g. Yu et al. 2002). The main classes of monomeric DON was approximated from seven soils (Warren 2013c). Note that data are averages and there is wide variation among soils, for example, protein amino acids accounted for between 52 and 88 % of monomers, quaternary ammonium compounds were 1-28 % of monomers, non-protein amino acids were 3–19 % of monomers, and other classes were 1–21 % of monomers
Size-based fractionation is one easy means of characterising DON. There is at least three orders of magnitude range in molecular weight of DON from the smallest organic compounds with molecular weights of less than 100 Da (e.g. ethanolamine, glycine, alanine) through to large polymers with molecular weights of 100s of kDa. Moreover, there are functional and ecological differences in DON molecules that are (directly or indirectly) related to molecular weight. For example, the < 1 kDa fraction of DON turns over rapidly (~hours) in comparison with the > 1 kDa fraction that turns over much more slowly (~days-months)(Jones et al. 2004). To date the most comprehensive study incorporating size-based fractionation reported that most DON in grasslands soils was in the > 100 kDa fraction, with the next largest fraction being < 1 kDa, with little in intermediate (1–100 kDa) fractions (Farrell et al. 2011a). A broadly similar trend was reported in a study examining dissolved organic carbon, with a larger proportion of DOC in the <1 kDa fraction than the 1–10 kDa fraction, but unfortunately no data were reported for larger size fractions (Marschner and Kalbitz 2003). The paucity of studies performing size-based fractionation precludes strong generalisations; nevertheless, it would appear that most DON is in molecules larger than 100 kDa and smaller than < 1 kDa, with a smaller proportion in 1–100 kDa fractions.
Is knowing compound classes sufficient?
For most compound classes we know astonishingly little about the individual compounds comprising the pool (Table 1), but do we really need detailed molecular information? In other words, is knowledge at the level of compound or molecular weight classes good enough? For many biological questions molecular information is required. The first reason is that there is molecular specificity of the transporters responsible for uptake and efflux of DON in microbes and plants. For example, plants take up different amino acids at different rates (Persson and Nasholm 2001a; Svennerstam et al. 2011). Similarly, based on experiments on mammals and microbes (Lister et al. 1995; Daniel et al. 2006) it is likely that uptake of peptides by plants and microbes varies depending on the peptide’s amino acid sequence. The second reason is that there is large variation among molecules in physico-chemical properties, even within compound and molecular weight classes. Notable among these properties are solubility and charge that affect interactions of molecules with the soil stationary phase. For example, acidic, neutral and basic amino acids diffuse through the soil at different rates (Owen and Jones 2001) and are differentially adsorbed to the soil stationary phase (Jones and Hodge 1999).
Molecular forms of organic N in soil solution: Free amino acids
Free amino acids tend to be the single most abundant class of organic N monomers in soil solution (Fig. 2). Widespread use of gas chromatography (GC) and high performance liquid chromatography (HPLC) derivatisation-based methods has led to a reasonable understanding of the patterns of primary amino acids, and to a lesser extent secondary amino acids (e.g. Read and Bajwa 1985; Abuarghub and Read 1988; Kielland 1995; Turnbull et al. 1996; Streeter et al. 2000; Andersson and Berggren 2005; Warren 2008; Werdin-Pfisterer et al. 2009; Jämtgård et al. 2010; Farrell et al. 2011a; Inselsbacher et al. 2011). It is rare for all the protein amino acids to be detected within one soil, but by combining data from multiple studies it is apparent that soil solution or soil extracts can contain all protein amino acids plus a few common non-protein amino acids such as GABA, citrulline and ornithine. Comparison of amino acid profiles among studies is generally not possible because analytical methods differ in which amino acids are quantifiable (some methods cannot quantify amides, some cannot quantify secondary amino acids, some cannot quantify cysteine or methionine) and because amino acid profiles of soil extracts (H2O or K2SO4 or KCl extracts) are not strictly comparable with amino acid profiles of soil solution (Jones and Willett 2006; Warren 2008).
Nevertheless if we restrict our analysis to one sample type—soil solution—it is apparent that the dominant amino acids vary among soils. For example, in soil solution of agricultural soils the dominant amino acids were glycine, serine, alanine, aspartic acid & asparagine and glutamic acid & glutamine (Jämtgård et al. 2010), while in grasslands they were glycine, lysine and threonine (Farrell et al. 2011a) and in forest soils were serine, glutamic acid, aspartic acid, leucine (Yu et al. 2002). Variation amongst sites in amino acid profiles has also been observed in studies involving soil solutions collected across natural gradients (Yu et al. 2002) or from diverse sites (Warren 2013c). Clearly, the gross differences in amino acid profiles reported within studies (Yu et al. 2002; Warren 2013c) cannot be due to methodological differences, and thus it would appear that differences among soils are real. This contrasts with some previous studies suggesting that the composition of the soil amino acid pool is affected little by soil type (Bremner 1966; Sowden et al. 1977; Stevenson 1982).
Few studies have distinguished between L- and D- enantiomers of amino acids, yet the distinction is non-trivial due to vast differences between enantiomers in patterns of uptake and metabolism (Bruckner and Westhauser 2003; Hill et al. 2011; Vranova et al. 2012). The occurrence and metabolism of D-amino acids in soils has been recently reviewed (Vranova et al. 2012), so only brief details are given here. Nineteen out of 20 protein amino acids can exist as L- or D-enantiomers (“mirror images“) because their alpha carbon molecules are bonded to four different groups (COOH, R, NH2 and H). Two of the protein amino acids (isoleucine, threonine) have two chiral carbons and thus four stereoisomers: two enantiomers (D- and L-isoleucine and D- and L-threonine) and two diastereoisomers (non-mirror image stereoisomers D- and L- allo-isoleucine and D- and L-allo-threonine). Glycine has two (indistinguishable) hydrogens attached to its alpha carbon and thus is achiral. D-amino acids are less abundant in nature than their corresponding L-enantiomers, yet D-amino acids have been reported from a range of taxa (Khoury et al. 2011) and in habitats such as soil (e.g. Amelung and Zhang 2001; Wichern et al. 2004). D-amino acids account for a significant but highly variable proportion of amino acids in hydrolysed soil (Pollock et al. 1977; Amelung and Zhang 2001; Amelung 2003; Wichern et al. 2004), but much less is known about their occurrence in the soil solution (Vranova et al. 2012). Aqueous ethanol extracts of soil from an orchard indicated that 2–17 % of amino acids were D-enantiomers (Bruckner and Westhauser 2003), while aqueous ethanol extracts of a peat bog indicated that the D/L ratio varied among the seven detected chiral amino acids (D/L between 40 and 1 % in 0–20 cm samples) and generally increased with depth (Kunnas and Jauhiainen 1993). Results of these studies need to be treated with caution because aqueous ethanol extracts a proportion of amino acids from living organisms and thus ethanol extracts do not strictly represent the soil solution (Vranova et al. 2012). Hence, a priority for future research should be determining the D/L ratio of amino acids in samples that are more representative of the soil solution (e.g. obtained by microdialysis, centrifugal extraction or lysimetry).
Molecular forms of organic N in soil: Other monomers
Soil and the soil solution has long been known to contain a diversity of organic N monomers in addition to common amino acids (e.g. non-protein amino acids, nucleic acids, ureides, quaternary ammonium compounds, purine and pyrimidine bases) (e.g. Schreiner 1912; Schreiner and Skinner 1912; Lathrop 1917; Aseeva et al. 1977; Aseeva et al. 1978; Schlimme and Kirse 1983). More recent studies using 15N NMR have indicated occurrence of a wealth of heterocyclic structures in fire-affected soils (Knicker et al. 2005; Knicker 2007; Knicker 2011). Unfortunately older studies were generally limited in scope presenting data for a handful of molecules and were qualitative or (at best) semi-quantitative, while more recent 15N NMR studies generally did not identify specific molecules but rather presence of functional groups. As a consequence it is not possible to draw together a cohesive picture of abundances and overall patterns from much of the literature. What one can do, however, is to piece together data from quantitative studies of particular compound classes (e.g. amino sugars: Roberts et al. 2007) with recent studies that used mass spectrometry to obtain a broader picture of organic N monomers (Warren 2013b; Warren 2013c).
Among seven soils protein amino acids were found to account for 52–88 % of organic N monomers (Warren 2013b; Warren 2013c), in broad agreement with views that amino acids are the major pool of organic N monomers in the soil solution (Jones et al. 2005). However, N-containing organic monomers from an additional 11 compound classes were quantified with the most abundant other compound classes being quaternary ammonium compounds and non-protein amino acids (Fig. 2). Moreover, only in one of seven soils did protein amino acids comprise all ten of the most abundant molecules. The more common situation was for the group of ten most abundant molecules to include quaternary ammonium compounds (e.g. betaine, hercynine), or non-protein amino acids (e.g. theanine) or pyrimidines (ectoine and hydroxyectoine) or azoles (urocanic acid) or alkylamines (triethanolamine).
Amino sugars have received less attention than amino acids, despite studies suggesting that amino sugars account for 5–12 % of total soil organic N (Stevenson 1983; Jorgensen and Richter 1992; Knicker 2011) and are cycled very rapidly in soils (Roberts et al. 2007; Roberts and Jones 2012). Part of the reason for the relative neglect of amino sugars is that concentrations in the soil solution tend to be rather small (individual amino sugars < 150 nM: Roberts et al. 2007) and make a much smaller contribution to DON than amino acids (Roberts et al. 2007; Warren 2013c). To date research on amino sugars in the soil solution has focussed on hexosamines such as glucosamine, yet it is probable that soil solution contains a far richer assemblage of amino sugars and molecules related to amino sugars (Table 1). The amino sugars in soil probably originate from breakdown of cell walls of plants, archea, bacteria and fungi (Roberts et al. 2007; Roberts and Jones 2012). Hence, soil solution could well contain other sugar amines derived from cell walls of archea, bacteria and fungi such as N-acetylglucosamine, N-acetylmuramic acid, N-acetyltalosaminuronic acid and their corresponding non-acetylated forms.
Molecular forms of organic N in soil: oligomers and polymers
Two independent techniques, hydrolysis (Yu et al. 2002; Andersson and Berggren 2005; Farrell et al. 2011b; Hill et al. 2011; Hill et al. 2012) and 15N NMR (Michalzik and Matzner 1999; Kögel-Knabner 2006) suggest that DON is dominated by oligomers and polymers of amino acids (i.e. peptides and proteins, see Fig. 2). For example, in a series of forest soils 48 to 74 % of DON could be accounted for by compounds that hydrolysed to amino acids (Yu et al. 2002). To date the focus has been on simple peptides and proteins (i.e. polymers of amino acids), but it is probable that soil solution contains other N-containing oligomers and polymers. For example, glyco-peptides are common constituents of cell walls of plants and bacteria and thus probably occur in the soil solution. More generally, the soil probably contains a diversity of conjugated or modified peptides and proteins (e.g. glyco-, phospho-, lipo-, chromo- peptides and proteins) given that such modifications of proteins and peptides are common (Mann and Jensen 2003; Blom et al. 2004; Khoury et al. 2011). Conjugation can have profound effects on physico-chemical properties and metabolism of peptides and proteins, and thus a priority for future research should be to quantify separately conjugated and simple forms and investigate differences in metabolism (e.g. uptake by plants and microbes).
There have been few measurements of molecular forms of peptides and proteins, despite the recognition that they dominate DON and may make a significant contribution to ecosystem N cycling and plant nutrition (Yu et al. 2002; Andersson and Berggren 2005; Rentsch et al. 2007; Paungfoo-Lonhienne et al. 2008; Hill et al. 2011; Soper et al. 2011; Hill et al. 2012; Paungfoo-Lonhienne et al. 2012). The first (and apparently only) study so far to profile peptides in the soil solution reported that the soil solution contained at least several hundred peptides < 1 kDa (Warren 2013a). The large number of peptides meant that the pool of peptide-N was spread among many molecules. Hence, the most abundant molecules were amino acids and quaternary ammonium compounds with individual concentrations of 0.5–1.0 μmol L−1, whereas approximate concentrations of the most abundant peptides were 0.01–0.05 μmol L−1.
Recent studies suggest that chirality may have a large effect on uptake of peptides from soil (Hill et al. 2011; Hill et al. 2012), yet there are currently no data on chirality of peptides (or proteins) in the soil solution and the situation is enormously complex. For example, if we ignore chirality there are 3.3684 × 106 possible amino acid sequences for di- to penta-peptides (Payne et al. 2000). The broader literature on peptide transporters indicates that uptake parameters vary among the different sequence combinations of D- and L- amino acids (e.g. di-peptides that are LL, DD, DL, LD)(Lister et al. 1995). Thus the complexity arising from variation in amino acid sequence is multiplied by the occurrence of stereoisomers.
Sampling of organic N from soil solution
The common methods for characterising DON involve sub-sampling the soil solution (centrifugation, microfiltration via suction lysimeters), or dialysing analytes from the soil solution (microdialysis), or extracting bulk soil with aqueous solutions. A germane question to ask is whether the different sampling/extraction approaches yield samples that reflect in situ concentrations. One way to evaluate sampling/extraction approaches would be to compare them with concentrations measured directly in situ, for example, by mass spectrometry surface analysis methods (see below). Comparisons such as these are yet to be made, and thus we are left in the unsatisfactory position of knowing results differ significantly among sampling/extraction approaches (e.g. Jones and Willett 2006; Inselsbacher et al. 2011) but cannot objectively say which methods yield results closest to in situ concentrations. Bearing this important caveat in mind, what follows is a brief discussion of the more common methods to sample organic N from the soil solution.
Soil biologists have long used microfiltration-based suction lysimeters for sampling of the soil solution and characterisation of DON (Yu et al. 2002; Jämtgård et al. 2007; Inselsbacher et al. 2011). A microfiltration probe (commonly referred to as a suction lysimeter, porous cup or porous probe) contains a hydrophilic semi-permeable membrane that is buried in the ground and is connected to the surface by vacuum tubing. Application of a partial vacuum leads to mass flow of soil solution (the filtrate) into the probe lumen and a collection device (Fig. 3). In general, lysimeters involve filtration through small pores (< 0.1 or 0.2 μm) so as to exclude microbes and thus the filtrate should be sterile and not suffer rapid mineralisation after collection. The amount of disturbance of the soil varies depending on the size of the microfiltration probe and rate at which soil solution is withdrawn. Ideally one would use the smallest possible probe (e.g. <1 mm diameter probes used in neuroscience) and withdraw soil solution slowly so as not to affect the external milieu. In practice, however, the microfiltration probes commonly used by soil biologists tend to be larger (e.g. the smallest commercially available “micro” probe is 1 mm diameter and most probes are > > 2.5 mm) and involve rapid depletion of the external milieu due to removal of large volumes of soil solution (100s of μL to mL). Microfiltration is typically regarded as providing representative samples because it is essentially a size-based filtration process (Korf et al. 2010), but samples of soil solution obtained via microfiltration can be biased for at least two reasons. First, filtrates obtained from soil are probably biased towards the largest liquid-filled pores because pores tend to empty from largest to smallest as a function of trends in water potential (Miro et al. 2010). The second reason is that some molecules can be adsorbed to microfiltration membranes (e.g. up to 50 % of an artificial DOC solution was adsorbed by ceramic suction cups, Wessel-Bothe et al. 2000), and there is some selectivity in terms of the rate at which different molecules cross the membrane. Adsorption and relative permeability is a function of the materials membranes are made from (borosilicate, silicon carbide, quartz/teflon, ceramic, polyethersulfone, regenerated cellulose) and varies among molecules (e.g. as a function of charge or hydrophilicity) (Guggenberger and Zech 1992; Murphy et al. 2000; Wessel-Bothe et al. 2000; Huinink et al. 2010; Korf et al. 2010). Hence, absolute quantification requires testing of recovery of target molecules. A major limitation of microfiltration is that soils must be moist so as to obtain soil solution (e.g. > 70 % of maximum water holding capacity: Miro et al. 2010).

Schematic illustration of the processes of microdialysis and microfiltration
Centrifugal extraction of soil solution (Davies and Davies 1963; Monreal and Mcgill 1985; Zabowski 1989) operates via a similar principle as microfiltration: soil solution is extracted from soil by application of a pressure differential across a semi-permeable membrane. Centrifugal extraction, like microfiltration, is complicated by differences among molecules in relative permeability across the semi-permeable membrane and is probably biased towards the easily extracted fraction of soil solution (i.e. largest water-filled pores). A major drawback of centrifugal extraction is that it involves disruption of soil structure and delays in-between soil collection and extraction. Metabolism of analytes can occur at any of the steps in between sample collection and extraction, and thus concentrations measured in centrifugal extracts may deviate from in situ concentrations.
Microdialysis has recently been used as a tool to monitor in situ diffusive flux of amino acids in soils (Inselsbacher et al. 2011; Inselsbacher and Näsholm 2012), which followed on from earlier work on microdialysis in soil (Miro and Frenzel 2005; Sulyok et al. 2005) and several decades experience by the neuroscience community (e.g. as reviewed by: Korf et al. 2010). Microdialysis involves insertion of a small probe into the soil and subsequent perfusion of the probe lumen with a perfusate solution (Fig. 3). The probe contains a semi-permeable membrane that allows small molecules to penetrate the membrane down a concentration gradient (Miro and Frenzel 2005). The outflow from the probe (dialysate) contains the analytes of interest and can subsequently be analysed (Korf et al. 2010). As is the case for microfiltration, relative permeability of the microdialysis membrane varies among molecules, and thus the material of the membrane must be chosen wisely and various procedures used to derive the tissue concentration of an analyte from its concentration in the dialysate (i.e. calibration or calculation of relative recovery, e.g. Bungay et al. 1990; Menacherry et al. 1992; Bouw and Hammarlund-Udenaes 1998). One advantage of microdialysis over microfiltration is that microdialysis can obtain samples from dryer soil than microfiltration (minimum of 50 % versus 70 % of maximum water-holding capacity: Miro et al. 2010). To place this in context, in most soils the notional permanent wilting point (−1.5 MPa) equates to considerably less than the 50 % of maximum water holding capacity stated as the lower limit of operation for microdialysis. Hence, for a drying soil microdialysis will stop working well before plants stop extracting water and nutrients.
The Achilles heel of microdialysis is that chemical composition of the perfusate and sample matrix (i.e. soil) affect microdialysis such that dialysate concentrations of ionic compounds may vary for the same concentration of analyte (Miro and Frenzel 2004; Miro and Frenzel 2005). This complication arises because of differences of migration rates between the target ionic compound and the concomitant co-ions. Solutions include matching perfusate composition with that of the sample matrix and/or using perfusates containing substantial concentrations of inorganic salts. For neurochemical and clinical applications choice of perfusate composition is comparatively simple because the inorganic salt composition and ionic strength of the sample matrix is rather invariant; hence, the common practice is to use perfusates that are isotonic solutions of inorganic salts. Recent papers on soil microdialysis used perfusate solutions consisting solely of water (Inselsbacher et al. 2011; Inselsbacher and Näsholm 2012), which would tend to bias microdialysis towards uncharged compounds because their mass transfer is unaffected by co-ions (Miro and Frenzel 2004; Miro and Frenzel 2005). The relative permeability of the probe towards different compounds can in principle be accounted for by calibration, but calibrations will be skewed if inorganic salt composition or ionic strength of the sample matrix is different to that used for calibration. This poses a major challenge for use of microdialysis with soil given that the inorganic salt composition of soil solution varies enormously among soils and is highly dynamic. Additional validation of microdialysis is required to determine if perfusate composition needs to be modified and calibrations of relative recovery (for 10s to 100 s of compounds) performed whenever there are changes in inorganic salt composition or ionic strength of the soil solution being probed.
Aqueous extracts are an easy means of obtaining a large amount of sample, and are the only technique that works reliably in dry soils. The main problems with soil extracts is that they involve disturbance of the soil, delays in-between collection and extraction, and extracts are “polluted” by a variable (and generally unknown) contribution of compounds extracted from lysed microbial and plant cells. Mineralization during extraction probably means that extracts overestimate concentrations of inorganic N and underestimate organic N (Rousk and Jones 2010). Additional complications are that the profile and amount of metabolites extracted are strongly dependent on the type of extractant (e.g. water versus 1 M KCl, Warren and Taranto 2010), shaking time and ratio of soil to extractant (Jones and Willett 2006). The limitations of soil extracts mean that they are unlikely to be representative of the in situ milieu, but yield data that can be compared within studies and (with rigorous control of extraction conditions) among studies. Moreover, extracts may be the only viable option in some situations such as in dry soil or where a large volume of sample is required for subsequent analyses.
Analytical approaches for quantifying organic N in soil: Molecular weights and compound classes
Our views of DON are shaped and limited by the analytical approaches used. Thus it is necessary to evaluate critically the methods used for analysing DON and suggest what might be fruitful approaches to pursue in the future. To review all of the methods used to explore DON is impractical, so instead I focus on those that are used most commonly and/or have the most potential for future studies. I begin by considering methods for quantifying broad pools of DON, and then move on to consider methods for uncovering the molecular nature of DON.
DON comprises multiple compound classes (Schreiner 1912; Schreiner and Skinner 1912; Lathrop 1917; Dadd et al. 1953; Aseeva et al. 1977; Aseeva et al. 1978; Schlimme and Kirse 1983; Warren 2013b; Warren 2013c) and thus one way to characterise DON is to quantify total pools of different compound classes. To date the most common of these measurements has been of the total pool of amino acids estimated from ninhydrin (colourimetric) or o-phthaldialdehyde (fluorometric) assays (Jones et al. 2002). An important caveat with ninhydrin and o-phthaldialdehyde assays is that they are neither completely specific nor comprehensive for amino acids. The assays are based on the reaction of ninhydrin with primary and secondary amines to produce coloured derivatives (λmax = 570 nm for primary amines and 440 nm for secondary amines), and reaction of o-phthaldialdehyde with primary amines and sulfhydryls to produce a fluorescent derivative. In practice secondary amines such as proline (which can be abundant in many soils: Warren 2013c) are generally not captured by the ninhydrin method because measurements are typically made only at 570 nm. In both assays the degree of colour or fluorescence development depends on the individual amino acid (Jones et al. 2002) and both assays suffer from interferences (e.g. of ninhydrin with ammonium and to a lesser extent peptides and proteins; and o-phthaldialdehyde with sulfhydryls). Modifications of the classical methods have been published that eliminate some interference problems (e.g. carbohydrate interference with ninhydrin: Magné and Larher 1992), yet these methods do not seem to be widely used. A consequence of these limitations is that the assays can be quantitative, but only after extensive testing and correction for cross-reactivity. For many studies it is unclear if these tests have been performed and consequently data should be interpreted cautiously. Colorimetric assays exist for other compound classes such as amino sugars (Reissig et al. 1955), purines (Schlimme and Kirse 1983) and quaternary ammonium compounds (Stumpf 1984) but do not seem to have been widely used for soil extracts or soil solution. Colourimetric and fluorimetric assays have also been used to estimate total pool of proteins, but the weight of evidence suggests that these assays are unreliable when used with crude samples of soil solution due to interferences and cross-reactivity issues (Whiffen et al. 2007; Roberts and Jones 2008).
The contribution of peptides and proteins to DON is commonly estimated by hydrolysing samples and then measuring released amino acids. Hydrolysis with 6 M hydrochloric acid is the most popular method, despite a litany of problems: poor recoveries of Trp, Cys, Ser, and Thr due to partial decomposition; poor recovery of Ile and Val due to slow cleaving of peptide bonds; poor recovery of Met due to oxidation; and hydrolysis of Asn and Gln to Asp and Glu (Rutherford and Gilani 2009). Alternatives such as hydrolysis with methanesulfonic acid have been used with soils and offer advantages and disadvantages over the more usual 6 M hydrochloric acid (Martens and Loeffelmann 2003). In reality there is no best method and it seems unlikely that such a “silver bullet” will arise given that the commonly-used hydrolysis procedures have changed little in 50 years despite large numbers of papers being published on the topic (Rutherford and Gilani 2009). The most important aspect of hydrolysis is knowing limitations of the approach and implications for data interpretation. In terms of knowing the fraction of DON in peptides and proteins, results of hydrolysis have a wide error margin (maybe ± 10–20 %) due to poor recoveries of several amino acids and because Asp and Glu quantified in the hydrolysate are the sum of the acid and amide (that differ by 1 N). Moreover, the products of hydrolysis are usually interpreted in terms of simple peptides and proteins, yet in reality are probably a mix of simple and conjugated forms (e.g. glyco-, phospho-, lipo- peptides & proteins).
One way to partially characterise DON is based on molecular weight. The simplest approach is to fractionate samples with membranes of differing molecular weight cut-offs, and perform subsequent measurements on the fractionated sample (e.g. Farrell et al. 2011a). Automated on-line systems for size based separations do not appear to have been used for analysis of soil solution, but could provide some useful information. For example, it is rather commonplace for marine and freshwater dissolved organic matter to be characterised with on-line systems involving size exclusion chromatography coupled with multiple on-line detectors (ultra-violet, fluorescence and dissolved organic carbon) (Her et al. 2003).
Analytical approaches for quantifying organic N in soil: Molecular information
The number and diversity of organic N molecules in the soil solution poses a considerable analytical challenge. For example, recent studies with capillary electrophoresis-mass spectrometry (CE-MS) suggested there were at least 100 monomeric DON compounds and several hundred small peptides (Warren 2013b; Warren 2013c; Warren 2013a) while direct infusion ultra-high resolution mass spectrometry of aqueous extracts resolved thousands of signals from organic compounds (Ohno et al. 2010). The enormous range in size, solubility, polarity and volatility means that it is not possible to capture the molecular diversity of DON with one technique. For biological samples the problem of extreme chemical diversity is rather commonplace and commonly requires use of multiple complementary analytical platforms to obtain a broad overall picture (Mashego et al. 2007; Saito et al. 2010; Patti et al. 2012; Kuehnbaum and Britz-McKibbin 2013).
In deciding how one might analyse DON in soil solution it is worth thinking about the biggest hurdles to overcome. For at least three reasons the biggest challenge for analysing DON in soil solution is detection and identification.
-
1.
Few DON compounds have concentrations > 1 μM and thus detection limits need to be much better than 1 μM to be useful for DON in soil solution.
-
2.
Detection needs to be able to cope with complex mixtures containing hundreds to thousands of individual compounds. The large number of compounds in DON means that there will be multiple overlapping peaks if samples are separated using common analytical separations such as GC, HPLC or CE. Non-selective (“universal”) detectors such as ultraviolet–visible detector (UV–vis), evaporative light-scattering detector (ELSD) and flame ionization detector (FID) can visualise peaks but provide little structural information and thus are unable to distinguish overlapping peaks and cope with complex mixtures.
-
3.
Many DON compounds in the soil solution are unknowns. An ideal analytical platform should involve detection methods that can aid with ID (i.e. yield structural information).
The two main detection systems capable of ID and quantification of complex mixtures are nuclear magnetic resonance spectroscopy (NMR) and mass spectrometry (MS). Both are considered briefly below.
Nuclear magnetic resonance spectroscopy is the primary analytical tool for determining structure of molecules, and can also be used for ID and quantification of small molecules in complex mixtures (Nicholson et al. 1999; Fan et al. 2001; Dunn et al. 2011; Kuehnbaum and Britz-McKibbin 2013). The most common type of NMR used in studies on DON has been solid-state 15N or 13C NMR, which has provided information about presence and relative abundance of particular functional groups of N (e.g. Knicker et al. 1995; Knicker and Ludemann 1995; Knicker et al. 1996; Schmidt et al. 1997; Chen and Xu 2008) rather than ID of individual compounds. One limitation of NMR for characterising DON in soil solution is that NMR has limited concentration sensitivity. Choice of instrumental approach is important because solid-state NMR tends to be more sensitive than liquid state NMR, while 13C is more sensitive than 15N (Mopper et al. 2007). In any case, the problem of poor detection limits can be partially overcome by pre-concentration of samples (e.g. by freeze drying or solid-phase extraction), isotope labelling and longer acquisition times. Future applications of NMR are likely to involve advanced multi-dimensional techniques and spin editing, as has been suggested for marine dissolved organic matter (Mopper et al. 2007; Hertkorn et al. 2013).
Mass spectrometry is in general more sensitive than NMR, but the different types of mass spectrometers and various ways they can be used lead to great differences in detection limits from better than 1 nM to low μM (Lu et al. 2008; Psychogios et al. 2011; Warren 2013b). Mass spectrometers can be used as stand-alone instruments whereby the sample is infused directly into the mass spectrometer (commonly known as direct infusion or flow injection mass spectrometry). Direct-infusion mass spectrometry has been widely used for high-throughput phenotyping of plant and animal samples (Dunn et al. 2011; Draper et al. 2013), but has seen infrequent use with soil. The biggest challenge with direct infusion mass spectrometry is resolving individual compounds within complex mixtures containing several thousand individual compounds, including > 60 peaks per nominal mass (Koch et al. 2005; Purcell et al. 2006; Ohno et al. 2010). Low-resolution mass spectrometers (e.g. ion trap, quadrupole and triple quadrupole) can resolve ions differing in nominal mass (e.g. glutamine with nominal mass 146 Da from glutamate with nominal mass 147 Da) whereas ions with same nominal mass but differing exact mass (e.g. glutamine with exact mass 146.06859 Da versus lysine with exact mass 146.10498 Da) are not resolved from one another and appear as a single peak. Consequently, with direct infusion of complex samples, low-resolution mass spectrometers have limited ability to resolve and identify individual compounds– unless additional structural information is obtained by, for example, obtaining MS2 and MS3 spectra (Koulman et al. 2007). In contrast, high and ultra-high resolution mass spectrometers (e.g. Fourier transform-ion cyclotron resonance FT-ICR, and Orbitrap) can resolve ions differing in mass by less than 0.001 Da even with extremely complex spectra (Kaiser et al. 2011). Moreover, high and ultra-high resolution mass spectrometers yield accurate molecular weight and isotope pattern information that can be used to predict the elemental compositions (molecular formulas) of metabolites as clues for their structural elucidation. Direct infusion experiments with high or ultra-high resolution mass spectrometers have provided enormous detail on the composition of thousands of individual compounds in dissolved organic matter (DOM) from freshwater and marine systems (Stenson et al. 2003; Kujawinski et al. 2004; Koch et al. 2005; Hertkorn et al. 2013). In one of the few applications of direct infusion mass spectrometry on soil, Ohno and colleagues (2010) used direct infusion with ultra-high resolution mass spectrometry of plant and soil extracts to discover molecular signals (“biomarkers”) for different types of soil and plant-derived biomass.
Mass spectrometry surface analysis methods such as DESI (desorption electrospray ionization)(Takáts et al. 2004) hold great promise as tools for direct chemical analysis of soil. In DESI an electrically charged mist is directed at the sample surface, analytes are desorbed and ions travel into the mass spectrometer. DESI and related methods permit the direct chemical analysis of surfaces at high spatial and temporal resolution, require no sample preparation, operate in ambient air rather than vacuum and can be applied to live samples (Takáts et al. 2005). Mass spectrometry surface analysis methods are yet to be applied to soil, but a miniaturised version of DESI (nanoDESI) was recently used for metabolic profiling of living microbial colonies (Moree et al. 2012; Watrous et al. 2012) and can be applied to various other biological surfaces (e.g. see recent review of microbiological applications: Watrous and Dorrestein 2011). Some method development will be required to adapt surface analysis methods to soil, but the effort should be worthwhile given the potential reward is a method that meets many of the criteria of an ideal method: direct analysis, minimally invasive, high spatial and temporal resolution.
Where the aim is to ID and quantify as many compounds as possible in complex mixtures the most promising approach is chromatographic or electrophoretic separations hyphenated with mass spectrometry (Kuehnbaum and Britz-McKibbin 2013). In this approach crude sample is first separated by gas chromatography (GC), high performance liquid chromatography (HPLC) or capillary electrophoresis (CE) with the separated sample components subsequently entering a mass spectrometer. Major advantages of chromatographic or electrophoretic separations compared with direct infusion are: a) ions with the same nominal mass but different exact mass, which in direct infusion experiments with low-resolution mass spectrometers cannot be resolved, can often be separated by electrophoresis or chromatography; and b) structural isomers, which confound interpretation on any mass spectrometer, can often be separated by electrophoresis or chromatography.
GC-MS is the most mature hyphenated separation technique and has found widespread use for complex samples of biological origin (Roessner et al. 2000; Persson and Nasholm 2001b; Schauer et al. 2005; Wishart et al. 2009). The Achilles heel of GC is that analytes must be volatile to be chromatographed, which in practice means that GC-MS is limited to small molecules that are volatile or can be made volatile by derivatizing. However, many organic N molecules (e.g. quaternary ammonium compounds, oligomers and polymers) cannot be derivatized using common reagents or are degraded during passage through the hot injection port and column or have too low a vapour pressure (Kaspar et al. 2009). Hence, GC-MS is limited in scope to a sub-set of monomeric DON compounds (amino acids, some amines).
Mass spectrometry hyphenated with HPLC (LC-MS) or CE (CE-MS) is ideal for quantifying and identifying compounds comprising DON. To date LC-MS has not been applied to analysis of DON in soil solution, despite successful use in other biological systems for similarly complex samples. A useful starting point for LC-MS of small (< 1 kDa) molecules would be to use a combination of hydrophilic interaction liquid chromatography (HILIC) and reversed-phase separations to obtain a broad coverage of hydrophilic and hydrophobic molecules (Gika et al. 2008; Chen et al. 2012; Zhang et al. 2012). Recently CE-MS was used successfully for ID and quantification of DON monomers and small peptides in the soil solution (Warren 2013b; Warren 2013c; Warren 2013a). The CE-MS method was somewhat selective for DON because it separated those molecules that were cations at low pH, which in the case of organic molecules is typically associated with the presence of N (or less commonly other elements such as S or Cl). The CE-MS method could not separate DON molecules that were neutral or anionic at low pH such as N-acetylglucosamine and nucleoside mono- di- and tri-phosphates. However, these molecules are in principle separable using CE-MS approaches for anions (Soga et al. 2009; Kok et al. 2011).
The reality is that there is no single best analytical platform for DON in soil solution, and the best solution is to use multiple orthogonal separation and detection schemes. One aspect that has not been addressed here, yet arguably comes closest to being a “silver bullet” is use of NMR as a chromatographic detector or parallel collection of mass spectrometry and NMR data. In reality mass spectrometry yields enough structural information for ID of linear macromolecules with a limited number of building blocks (e.g. peptides: Hunt et al. 1986), but is often incapable of providing sufficient information for ID of general unknowns (Sturm and Seger 2012). LC–NMR combines efficient HPLC separation of crude mixtures with the information rich spectroscopic method NMR and effectively fills the sensitivity gap between highly sensitive LC–MS and the (typically) poor sensitivity of off-line NMR spectroscopy. LC-NMR and LC-SPE-NMR technologies are nowadays reasonably well established (Simpson et al. 2004; Godejohann 2010; Sturm and Seger 2012), yet the technology is not widespread and is yet to be applied to soil solution. An additional development that may well become the gold standard is the so-called “total analysis system” that separates samples by HPLC and then collects in parallel information rich NMR and MS signals (i.e. LC-SPE-NMR-MS: Schlotterbeck and Ceccarelli 2009).
Generating hypotheses from knowledge of the molecular composition of DON
Knowledge of the molecular composition of DON provides a solid foundation for generation of hypotheses that can be tested in future studies. What are some of the open questions or hypotheses arising from a broader and more inclusive understanding of DON?
Are the large amounts of non-amino acid monomers (e.g. ectoine, hydroxyectoine and quaternary ammonium compounds such as betaine and hercynine, Fig. 2) important for plant N nutrition? These molecules are suggested to be osmolytes of microbial and/or plant origin (Lippert and Galinski 1992; Hasegawa et al. 2000; Wood et al. 2001), but almost nothing is known about their cycling. It is likely that cycling of osmolytes would contrast with protein amino acids given their differing origins (de novo synthesis of osmolytes versus hydrolysis of proteins) and functions. It is known that bacteria can take up pre-formed osmolytes from the external medium (Welsh 2000; Wood et al. 2001) but we do not know if plants are also able to take up non-amino acid monomers and how this compares to rates of uptake of amino acids? Related questions that arise are whether re-wetting of dry soils lead to a pulse of osmolytes into the external medium due to osmotic de-shocking of microbes, and are these osmolytes rapidly taken up by plants or microbes?
Questions about the significance of peptides for plant nutrition arise from the observation that the pool of peptides in soil is large, but is spread among many compounds and thus individual concentrations are low. Little is known about the molecular specificity and kinetics of peptide uptake by plants, but this is critical for interpreting the significance of peptides in soil. At present we do not know whether individual peptides compete for uptake, or are taken up independently. From the plant’s perspective is there a single large pool of peptides, or individual peptides present at low concentrations? If, as has been shown for mammals and microbes (Lister et al. 1995; Daniel et al. 2006), affinity varies as a function of amino acid sequence, do differences in affinity correlate with concentrations of individual peptides in the soil solution?
Conclusions
That organic N is important and should be studied is beyond contention, but by focusing on amino acids and their polymers have we lost focus of the big picture? One of the most widely heard justifications for studying organic N is that nitrate and ammonium constitute only a small proportion of bio-available soluble N, and thus focusing on nitrate and ammonium yields a biased view of the N cycle. Examining amino acids and their polymers is a step in the right direction in terms of obtaining a more realistic view of the N cycle. However, the soil solution contains many other classes of organic N. For example, the presence of heterocyclics and osmolytes provides clues as to soil history and function (i.e. fire and abiotic stresses), and direct evidence of a substantial fraction of N being diverted away from amino acids and their polymers. Priorities for future research include determining the broader implications of heterocyclics and osmolytes for the N cycle. Studies of these and other classes of organic N are necessary not just because they are interesting in their own right, but to place studies of amino acids and their polymers in a broader context.
Quantification and characterisation of organic N may at first seem like an insurmountable task, but is achievable if the task is broken down into smaller pieces and prioritised. One way to prioritise and fast-track improved understanding is by examining first those classes of molecules and/or processes that are either completely unknown or putatively important for the N cycle.
-
Determining stereochemistry of amino acids and peptides in the soil solution should be priorities for future research. Measurements are necessary because experimental evidence indicates large differences in metabolism of D- and L-enantiomers of amino acids and peptides, yet we do not know what is in the soil solution.
-
A priority for future research is determining the diversity of oligomers and polymers in the soil solution. Oligomers and polymers are generally interpreted as simple linear sequences of amino acids, yet it is probable that other classes such as phospho- and glyco-peptides are abundant. It is necessary to differentiate among oligomers and polymers in contrasting chemical classes because they may have very different function and behaviour in the soil. For example, compared with simple peptides and proteins, the presence of phosphorus and carbohydrate groups in phospho- and glyco-peptides drastically alters physico-chemical properties (e.g. charge and solubility) and by virtue of altering C:N:P ratios may also affect how molecules are metabolised (e.g. depending on whether microbes are C, N or P limited).
References
Abuarghub SM, Read DJ (1988) The biology of mycorrhiza in the ericaceae.12. Quantitative-analysis of individual free amino-acids in relation to time and depth in the soil-profile. New Phytol 108:433–441
Amelung W (2003) Nitrogen biomarkers and their fate in soil. J Plant Nutr Soil Sci-Z Fur Pflanzenernahrung Und Bodenkund 166:677–686
Amelung W, Zhang X (2001) Determination of amino acid enantiomers in soils. Soil Biol Biochem 33:553–562
Amundson R (2001) The carbon budget in soils. Annu Rev Earth Planet Sci 29:535–562
Andersson P, Berggren D (2005) Amino acids, total organic and inorganic nitrogen in forest floor soil solution at low and high nitrogen input. Water Air Soil Pollut 162:369–384
Anonymous 2006 World Population Prospects:the 2006 Revision. United Nations, New York.
Aseeva IV, Panikov NS, Chursina OT (1977) Content and composition of nucleic-acids in soddy podzolic soils. Pochvovedenie:85–91
Aseeva IV, Panikov NS, Chursina OT (1978) Quantitative determination of purine components of nucleic-acids in soddy podzolic soils. Pochvovedenie:60–69
Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649
Bouw MR, Hammarlund-Udenaes M (1998) Methodological aspects of the use of a calibrator in in vivo microdialysis - further development of the retrodialysis method. Pharm Res 15:1673–1679
Bremner JM (1966) Nitrogenous compounds. In: McLaren AD, Peterson GH (eds) Soil biochem. Marcel Dekker, New York, pp 221–232
Bruckner H, Westhauser T (2003) Chromatographic determination of L- and D-amino acids in plants. Amino Acids 24:43–55
Bungay PM, Morrison PF, Dedrick RL (1990) Steady-state theory for quantitative microdialysis of solutes and water in vivo and in vitro. Life Sci 46:105–119
Chapin FS, Moilanen L, Kielland K (1993) Preferential Use of organic nitrogen for growth by a nonmycorrhizal arctic sedge. Nature 361:150–153
Chen CRR, Xu ZHH (2008) Analysis and behavior of soluble organic nitrogen in forest soils. J Soils Sediments 8:363–378
Chen J, Zhou LN, Zhang XY, Lu X, Cao R, Xu CJ, Xu GW (2012) Urinary hydrophilic and hydrophobic metabolic profiling based on liquid chromatography-mass spectrometry methods: differential metabolite discovery specific to ovarian cancer. Electrophoresis 33:3361–3369
Christou M, Avramides EJ, Jones DL (2006) Dissolved organic nitrogen dynamics in a Mediterranean vineyard soil. Soil Biol Biochem 38:2265–2277
Christou M, Avramides EJ, Roberts JP, Jones DL (2005) Dissolved organic nitrogen in contrasting agricultural ecosystems. Soil Biol Biochem 37:1560–1563
Dadd CC, Fowden L, Pearsall WH (1953) An investigation of the free amino-acids in organic soil types using paper partition chromatography. J Soil Sci 4:69–71
Daniel H, Spanier B, Kottra G, Weitz D (2006) From bacteria to man: archaic proton-dependent peptide transporters at work. Physiology 21:93–102
Davies BE, Davies RI (1963) A simple centrifugation method for obtaining small samples of soil solution. Nature 198:216
Draper J, Lloyd A, Goodacre R, Beckmann M (2013) Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: a review. Metabolomics 9:4–29
Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL (2011) Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev 40:387–426
Fan TWM, Lane AN, Shenker M, Bartley JP, Crowley D, Higashi RM (2001) Comprehensive chemical profiling of gramineous plant root exudates using high-resolution NMR and MS. Phytochemistry 57:209–221
Farrell M, Hill PW, Farrar J, Bardgett RD, Jones DL (2011a) Seasonal variation in soluble soil carbon and nitrogen across a grassland productivity gradient. Soil Biol Biochem 43:835–844
Farrell M, Hill PW, Wanniarachchi SD, Farrar J, Bardgett RD, Jones DL (2011b) Rapid peptide metabolism: a major component of soil nitrogen cycling? Glob Biogeochem Cycles 25
Filley TR, Boutton TW (2006) Ecosystems in flux: molecular and stable isotope assessments of soil organic matter storage and dynamics. Soil Biol Biochem 38:3181–3183
Friedel JK, Scheller E (2002) Composition of hydrolysable amino acids in soil organic matter and soil microbial biomass. Soil Biol Biochem 34:315–325
Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai ZC, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA (2008) Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320:889–892
Gardenas AI, Agren GI, Bird JA, Clarholm M, Hallin S, Ineson P, Katterer T, Knicker H, Nilsson SI, Nasholm T, Ogle S, Paustian K, Persson T, Stendahl J (2011) Knowledge gaps in soil carbon and nitrogen interactions - from molecular to global scale. Soil Biol Biochem 43:702–717
Gika HG, Theodoridis GA, Wilson ID (2008) Hydrophilic interaction and reversed-phase ultra-performance liquid chromatography TOF-MS for metabonomic analysis of Zucker rat urine. J Sep Sci 31:1598–1608
Godejohann M (2010) LC-NMR/MS and LC-SPE-NMR/MS: technical realization, possibilities and limitations. Planta Med 76:WSV_1. doi:10.1055/s-0030-1264227
Gruber N, Galloway JN (2008) An earth-system perspective of the global nitrogen cycle. Nature 451:293–296
Guggenberger G, Zech W (1992) Sorption of dissolved organic-carbon by ceramic P-80 suction cups. Z Fur Pflanzenernahrung Und Bodenkund 155:151–155
Hasegawa PM, Bressan RA, Zhu JK, Bohnert HJ (2000) Plant cellular and molecular responses to high salinity. Annu Rev Plant Physiol Plant Mol Biol 51:463–499
Her N, Amy G, McKnight D, Sohn J, Yoon YM (2003) Characterization of DOM as a function of MW by fluorescence EEM and HPLC-SEC using UVA, DOC, and fluorescence detection. Water Res 37:4295–4303
Hertkorn N, Harir M, Koch BP, Michalke B, Schmitt-Kopplin P (2013) High-field NMR spectroscopy and FTICR mass spectrometry: powerful discovery tools for the molecular level characterization of marine dissolved organic matter. Biogeosciences 10:1583–1624
Hill PW, Farrell M, Jones DL (2012) Bigger may be better in soil N cycling: does rapid acquisition of small L-peptides by soil microbes dominate fluxes of protein-derived N in soil? Soil Biol Biochem 48:106–112
Hill PW, Quilliam RS, DeLuca TH, Farrar J, Farrell M, Roberts P, Newsham KK, Hopkins DW, Bardgett RD, Jones DL (2011) Acquisition and assimilation of nitrogen as peptide-bound and D-enantiomers of amino acids by wheat. Plos One 6
Huinink KD, Lambooij B, Jansen-van Zelm K, Cremers TIFH, van Oeveren W, Bakker PL, Venema K, Westerink BHC, Korf J (2010) Microfiltration sampling in rats and in cows: toward a portable device for continuous glucocorticoid hormone sampling. Analyst 135:390–396
Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR (1986) Protein sequencing by tandem mass-spectrometry. Proc Natl Acad Sci U S A 83:6233–6237
Hutchinson HB, Miller NHJ (1912) The direct assimilation of inorganic and organic forms of nitrogen by higher plants. J Agric Sci 4:282–U284
Inselsbacher E, Näsholm T (2012) The below-ground perspective of forest plants: soil provides mainly organic nitrogen for plants and mycorrhizal fungi. New Phytol 195:329–334
Inselsbacher E, Ohlund J, Jämtgård S, Huss-Danell K, Näsholm T (2011) The potential of microdialysis to monitor organic and inorganic nitrogen compounds in soil. Soil Biol Biochem 43:1321–1332
Jämtgård S, Näsholm T, Huss-Danell K (2007) Occurrence of amino acids in soil and their uptake in Barley. Amino Acids 33:221–231
Jämtgård S, Näsholm T, Huss-Danell K (2010) Nitrogen compounds in soil solutions of agricultural land. Soil Biol Biochem 42:2325–2330
Jones DL, Darrah PR (1993) Influx and efflux of amino-acids from Zea mays L roots and their implications for N-nutrition and the rhizosphere. Plant Soil 156:87–90
Jones DL, Healey JR, Willett VB, Farrar JF, Hodge A (2005) Dissolved organic nitrogen uptake by plants - an important N uptake pathway? Soil Biol Biochem 37:413–423
Jones DL, Hodge A (1999) Biodegradation kinetics and sorption reactions of three differently charged amino acids in soil and their effects on plant organic nitrogen availability. Soil Biol Biochem 31:1331–1342
Jones DL, Kielland K (2002) Soil amino acid turnover dominates the nitrogen flux in permafrost-dominated taiga forest soils. Soil Biol Biochem 34:209–219
Jones DL, Owen AG, Farrar JF (2002) Simple method to enable the high resolution determination of total free amino acids in soil solutions and soil extracts. Soil Biol Biochem 34:1893–1902
Jones DL, Shannon D, Murphy DV, Farrar J (2004) Role of dissolved organic nitrogen (DON) in soil N cycling in grassland soils. Soil Biol Biochem 36:749–756
Jones DL, Willett VB (2006) Experimental evaluation of methods to quantify dissolved organic nitrogen (DON) and dissolved organic carbon (DOC) in soil. Soil Biol Biochem 38:991–999
Jorgensen RG, Richter GM (1992) Composition of carbon fractions and potential denitrification in drained peat soils. J Soil Sci 43:347–358
Judd TS, Bennett LT, Weston CJ, Attiwill PM, Whiteman PH (1996) The response of growth and foliar nutrients to fertilizers in young Eucalyptus globulus (Labill.) plantations in Gippsland, southeastern Australia. For Ecol Manag 82:87–101
Kaiser NK, Quinn JP, Blakney GT, Hendrickson CL, Marshall AG (2011) A Novel 9.4 Tesla FTICR mass spectrometer with improved sensitivity, mass resolution, and mass range. J Am Soc Mass Spectrom 22:1343–1351
Kaspar H, Dettmer K, Chan Q, Daniels S, Nimkar S, Daviglus ML, Stamler J, Elliott P, Oefner PJ (2009) Urinary amino acid analysis: a comparison of iTRAQ (R)-LC-MS/MS, GC-MS, and amino acid analyzer. J Chromatogr B-Anal Techn Biomed Life Sci 877:1838–1846
Kell DB, Oliver SG (2004) Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. BioEssays 26:99–105
Khoury GA, Baliban RC, Floudas CA (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1
Kielland K (1995) Landscape patterns of free amino acids in arctic tundra soils. Biogeochemistry 31:85–98
Knicker H (2007) How does fire affect the nature and stability of soil organic nitrogen and carbon? A review. Biogeochemistry 85:91–118
Knicker H (2011) Soil organic N - an under-rated player for C sequestration in soils? Soil Biol Biochem 43:1118–1129
Knicker H, Almendros G, GonzalezVila FJ, Ludemann HD, Martin F (1995) C-13 and N-15 NMR analysis of some fungal melanins in comparison with soil organic matter. Org Geochem 23:1023–1028
Knicker H, Almendros G, GonzalezVila FJ, Martin F, Ludemann HD (1996) C-13- and N-15-NMR spectroscopic examination of the transformation of organic nitrogen in plant biomass during thermal treatment. Soil Biol Biochem 28:1053–1060
Knicker H, Gonzalez-Vila FJ, Polvillo O, Gonzalez JA, Almendros G (2005) Fire-induced transformation of C- and N-forms in different organic soil fractions from a dystric cambisol under a mediterranean pine forest (pinus pinaster). Soil Biol Biochem 37:701–718
Knicker H, Ludemann HD (1995) N-15 and C-13 CPMAS and solution NMR-studies of N-15 enriched plant-material during 600 days of microbial-degradation. Org Geochem 23:329–341
Koch BP, Witt MR, Engbrodt R, Dittmar T, Kattner G (2005) Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochim Et Cosmochim Acta 69:3299–3308
Kögel-Knabner I (2006) Chemical structure of organic N and organic P in soils. In: Nannipieri P, Smalla K (eds) Nucleic acids and proteins in soils. Springer, Berlin, pp 23–48
Kok MGM, de Jong GJ, Somsen GW (2011) Sensitivity enhancement in capillary electrophoresis-mass spectrometry of anionic metabolites using a triethylamine-containing background electrolyte and sheath liquid. Electrophoresis 32:3016–3024
Korf J, Huinink KD, Posthuma-Trumpie GA (2010) Ultraslow microdialysis and microfiltration for in-line, on-line and off-line monitoring. Trends Biotechnol 28:150–158
Koulman A, Tapper BA, Fraser K, Cao MS, Lane GA, Rasmussen S (2007) High-throughput direct-infusion ion trap mass spectrometry: a new method for metabolomics. Rapid Commun Mass Spectrom 21:421–428
Kranabetter JM, Dawson CR, Dunn DE (2007) Indices of dissolved organic nitrogen, ammonium and, nitrate across productivity gradients of boreal forests. Soil Biol Biochem 39:3147–3158
Kuehnbaum NL, Britz-McKibbin P (2013) New advances in separation science for metabolomics: resolving chemical diversity in a post-genomic era. Chemical Reviews
Kujawinski EB, Del Vecchio R, Blough NV, Klein GC, Marshall AG (2004) Probing molecular-level transformations of dissolved organic matter: insights on photochemical degradation and protozoan modification of DOM from electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Mar Chem 92:23–37
Kunnas AV, Jauhiainen TP (1993) Separation and identification of free amino-acid enantiomers in peat by capillary Gas-chromatography. J Chromatogr 628:269–273
Lathrop EC (1917) The organic nitrogen compounds of soils and fertilizers. J Frankl Inst 183:169–206
Lippert K, Galinski EA (1992) Enzyme stabilization by ectoine-type compatible solutes - protection against heating, freezing and drying. Appl Microbiol Biotechnol 37:61–65
Lister N, Sykes AP, Bailey PD, Boyd CAR, Bronk JR (1995) Dipeptide transport and hydrolysis in isolated loops of rat small-intestine - effects of stereospecificity. J Physiol-Lond 484:173–182
Lu W, Bennett BD, Rabinowitz JD (2008) Analytical strategies for LC-MS-based targeted metabolomics. J Chromatogr B-Anal Technol Biomed Life Sci 871:236–242
Magné C, Larher F (1992) High sugar content of extracts interferes with colorimetric determination of amino acids and free proline. Anal Biochem 200:115–118
Mann M, Jensen ON (2003) Proteomic analysis of post-translational modifications. Nat Biotechnol 21:255–261
Marschner B, Kalbitz K (2003) Controls of bioavailability and biodegradability of dissolved organic matter in soils. Geoderma 113:211–235
Martens DA, Loeffelmann KL (2003) Soil amino acid composition quantified by acid hydrolysis and anion chromatography-pulsed amperometry. J Agric Food Chem 51:6521–6529
Mashego MR, Rumbold K, De Mey M, Vandamme E, Soetaert W, Heijnen JJ (2007) Microbial metabolomics: past, present and future methodologies. Biotechnol Lett 29:1–16
Menacherry S, Hubert W, Justice JB (1992) Invivo calibration of microdialysis probes for exogenous compounds. Anal Chem 64:577–583
Michalzik B, Matzner E (1999) Dynamics of dissolved organic nitrogen and carbon in a central european norway spruce ecosystem. Eur J Soil Sci 50:579–590
Miro M, Fitz WJ, Swoboda S, Wenzel WW (2010) In-situ sampling of soil pore water: evaluation of linear-type microdialysis probes and suction cups at varied moisture contents. Environ Chem 7:123–131
Miro M, Frenzel W (2004) Investigation of chemical effects on the performance of flow-through dialysis applied to the determination of ionic species. Anal Chim Acta 512:311–317
Miro M, Frenzel W (2005) The potential of microdialysis as an automatic sample-processing technique for environmental research. Trac-Trends Anal Chem 24:324–333
Monreal CM, Mcgill WB (1985) Centrifugal extraction and determination of free amino-acids in soil solutions by Tlc using tritiated 1-fluoro-2,4-dinitrobenzene. Soil Biol Biochem 17:533–539
Mopper K, Stubbins A, Ritchie JD, Bialk HM, Hatcher PG (2007) Advanced instrumental approaches for characterization of marine dissolved organic matter: extraction techniques, mass spectrometry, and nuclear magnetic resonance spectroscopy. Chem Rev 107:419–442
Moree WJ, Phelan VV, Wu CH, Bandeira N, Cornett DS, Duggan BM, Dorrestein PC (2012) Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc Natl Acad Sci U S A 109:13811–13816
Murphy DV, Macdonald AJ, Stockdale EA, Goulding KWT, Fortune S, Gaunt JL, Poulton PR, Wakefield JA, Webster CP, Wilmer WS (2000) Soluble organic nitrogen in agricultural soils. Biol Fertil Soils 30:374–387
Näsholm T, Ekblad A, Nordin A, Giesler R, Högberg M, Högberg P (1998) Boreal forest plants take up organic nitrogen. Nature 392:914–916
Näsholm T, Kielland K, Ganeteg U (2009) Uptake of organic nitrogen by plants. New Phytol 182:31–48
Nicholson JK, Lindon JC, Holmes E (1999) ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29:1181–1189
Nordin A, Hogberg P, Näsholm T (2001) Soil nitrogen form and plant nitrogen uptake along a boreal forest productivity gradient. Oecologia 129:125–132
Ohno T, He Z, Sleighter RL, Honeycutt CW, Hatcher PG (2010) Ultrahigh resolution mass spectrometry and indicator species analysis to identify marker components of soil- and plant biomass-derived organic matter fractions. Environ Sci Technol 44:8594–8600
Owen AG, Jones DL (2001) Competition for amino acids between wheat roots and rhizosphere microorganisms and the role of amino acids in plant N acquisition. Soil Biol Biochem 33:651–657
Patti GJ, Yanes O, Siuzdak G (2012) Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 13:263–269
Paul JP, Williams BL (2005) Contribution of alpha-amino N to extractable organic nitrogen (DON) in three soil types from the scottish uplands. Soil Biol Biochem 37:801–803
Paungfoo-Lonhienne C, Lonhienne TGA, Rentsch D, Robinson N, Christie M, Webb RI, Gamage HK, Carroll BJ, Schenk PM, Schmidt S (2008) Plants can use protein as a nitrogen source without assistance from other organisms. Proc Natl Acad Sci U S A 105:4524–4529
Paungfoo-Lonhienne C, Visser J, Lonhienne TGA, Schmidt S (2012) Past, present and future of organic nutrients. Plant Soil 359:1–18
Payne JW, Grail BM, Marshall NJ (2000) Molecular recognition templates of peptides: driving force for molecular evolution of peptide transporters. Biochem Biophys Res Commun 267:283–289
Persson J, Nasholm T (2001a) Amino acid uptake: a widespread ability among boreal forest plants. Ecol Lett 4:434–438
Persson J, Nasholm T (2001b) A GC-MS method for determination of amino acid uptake by plants. Physiol Plant 113:352–358
Pollock GE, Cheng CN, Cronin SE (1977) Determination of D and L isomers of some protein amino-acids present in soils. Anal Chem 49:2–7
Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia JG, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS (2011) The human serum metabolome. Plos One 6
Purcell JM, Hendrickson CL, Rodgers RP, Marshall AG (2006) Atmospheric pressure photoionization fourier transform ion cyclotron resonance mass spectrometry for complex mixture analysis. Anal Chem 78:5906–5912
Read DJ, Bajwa R (1985) Some nutritional aspects of the biology of ericaceous mycorrhizas. Proc R Soc Edinb 85B:317–332
Reissig JL, Strominger JL, Leloir LF (1955) A modified colorimetric method for the estimation of N-acetylamino sugars. J Biol Chem 217:959–966
Rennenberg H, Dannenmann M, Gessler A, Kreuzwieser J, Simon J, Papen H (2009) Nitrogen balance in forest soils: nutritional limitation of plants under climate change stresses. Plant Biol 11:4–23
Rentsch D, Schmidt S, Tegeder M (2007) Transporters for uptake and allocation of organic nitrogen compounds in plants. FEBS Lett 581:2281–2289
Roberts P, Bol R, Jones DL (2007) Free amino sugar reactions in soil in relation to soil carbon and nitrogen cycling. Soil Biol Biochem 39:3081–3092
Roberts P, Jones DL (2008) Critical evaluation of methods for determining total protein in soil solution. Soil Biol Biochem 40:1485–1495
Roberts P, Jones DL (2012) Microbial and plant uptake of free amino sugars in grassland soils. Soil Biol Biochem 49:139–149
Roessner U, Wagner C, Kopka J, Trethewey RN, Willmitzer L (2000) Simultaneous analysis of metabolites in potato tuber by gas chromatography–mass spectrometry. Plant J 23:131–142
Rousk J, Jones DL (2010) Loss of low molecular weight dissolved organic carbon (DOC) and nitrogen (DON) in H2O and 0.5 M K2SO4 soil extracts. Soil Biol Biochem 42:2331–2335
Rutherford SM, Gilani GS (2009) Amino acid analysis. Curr Protoc Protein Sci. doi:10.1002/0471140864.ps1109s58
Saito N, Ohashi Y, Soga T, Tomita M (2010) Unveiling cellular biochemical reactions via metabolomics-driven approaches. Curr Opin Microbiol 13:358–362
Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K, Roessner-Tunali U, Forbes MG, Willmitzer L, Fernie AR, Kopka J (2005) GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett 579:1332–1337
Schimel JP, Bennett J (2004) Nitrogen mineralization: challenges of a changing paradigm. Ecology 85:591–602
Schlimme E, Kirse M (1983) Enzymatic and colorimetric determination of purine nitrogen in soils. Zeitschrift Fur Pflanzenernahrung Und Bodenkunde 146:207–216
Schlotterbeck G, Ceccarelli SM (2009) LC-SPE-NMR-MS: a total analysis system for bioanalysis. Bioanalysis 1:549–559
Schmidt MWI, Knicker H, Hatcher PG, KogelKnabner I (1997) Improvement of C-13 and N-15 CPMAS NMR spectra of bulk soils, particle size fractions and organic material by treatment with 10% hydrofluoric acid. Eur J Soil Sci 48:319–328
Schreiner O (1912) The organic constituents of soils. Science 36:577–587
Schreiner O, Skinner JJ (1912) Nitrogenous soil constituents and their bearing on soil fertility. US Government Printing Office.
Simpson AJ, Tseng LH, Simpson MJ, Spraul M, Braumann U, Kingery WL, Kelleher BP, Hayes MHB (2004) The application of LC-NMR and LC-SPE-NMR to compositional studies of natural organic matter. Analyst 129:1216–1222
Smil V (2000) Feeding the world: A challenge for the 21st century. MIT Press, Cambridge
Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M (2009) Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal Chem 81:6165–6174
Soper FM, Paungfoo-Lonhienne C, Brackin R, Rentsch D, Schmidt S, Robinson N (2011) Arabidopsis and lobelia anceps access small peptides as a nitrogen source for growth. Funct Plant Biol 38:788–796
Sowden FJ, Chen Y, Schnitzer M (1977) Nitrogen distribution in soils formed under widely differing climatic conditions. Geochimica Et Cosmochimica Acta 41:1524–1526
Stenson AC, Marshall AG, Cooper WT (2003) Exact masses and chemical formulas of individual Suwannee river fulvic acids from ultrahigh resolution electrospray ionization fourier transform ion cyclotron resonance mass spectra. Anal Chem 75:1275–1284
Stevenson FJ (1982) Nitrogen in agricultural soils. American Society of Agronomy, Madison, Wisconsin, 940
Stevenson FJ (1983) Isolation and Identification of Amino-Sugars in Soil. Soil Sci Soc Am J 47:61–65
Streeter TC, Bol R, Bardgett RD (2000) Amino acids as a nitrogen source in temperate upland grasslands: the use of dual labelled (C-13, N-15) glycine to test for direct uptake by dominant grasses. Rapid Commun Mass Spectrom 14:1351–1355
Stumpf DK (1984) Quantitation and purification of quaternary ammonium-compounds from halophyte tissue. Plant Physiol 75:273–274
Sturm S, Seger C (2012) Liquid chromatography-nuclear magnetic resonance coupling as alternative to liquid chromatography-mass spectrometry hyphenations: Curious option or powerful and complementary routine tool? J Chromatogr A 1259:50–61
Sulyok M, Miro M, Stingeder G, Koellensperger G (2005) The potential of flow-through microdialysis for probing low-molecular weight organic anions in rhizosphere soil solution. Anal Chim Acta 546:1–10
Svennerstam H, Jämtgård S, Ahmad I, Huss-Danell K, Näsholm T, Ganeteg U (2011) Transporters in Arabidopsis roots mediating uptake of amino acids at naturally occurring concentrations. New Phytol 191:459–467
Takáts Z, Wiseman JM, Cooks RG (2005) Ambient mass spectrometry using desorption electrospray ionization (DESI): instrumentation, mechanisms and applications in forensics, chemistry, and biology. J Mass Spectrom 40:1261–1275
Takáts Z, Wiseman JM, Gologan B, Cooks RG (2004) Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306:471–473
Tilman D (1999) Global environmental impacts of agricultural expansion: The need for sustainable and efficient practices. Proc Natl Acad Sci U S A 96:5995–6000
Turnbull MH, Schmidt S, Erskine PD, Richards S, Stewart GR (1996) Root adaptation and nitrogen source acquisition in natural ecosystems. Tree Physiol 16:941–948
van Hees PAW, Jones DL, Finlay R, Godbold DL, Lundstomd US (2005) The carbon we do not see - the impact of low molecular weight compounds on carbon dynamics and respiration in forest soils: a review. Soil Biol Biochem 37:1–13
Virtanen AI, Linkola H (1946) Organic nitrogen compounds as nitrogen nutrition for higher plants. Nature 158:515–515
Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, Schlesinger WH, Tilman DG (1997) Human alteration of the global nitrogen cycle: sources and consequences. Ecol Appl 7:737–750
Vranova V, Zahradnickova H, Janous D, Skene KR, Matharu AS, Rejsek K, Formanek P (2012) The significance of D-amino acids in soil, fate and utilization by microbes and plants: review and identification of knowledge gaps. Plant Soil 354:21–39
Warren CR (2006) Potential organic and inorganic N uptake by six Eucalyptus species. Funct Plant Biol 33:653–660
Warren CR (2008) Rapid and sensitive quantification of amino acids in soil extracts by capillary electrophoresis with laser-induced fluorescence. Soil Biol Biochem 40:916–923
Warren CR (2013a) Development of a capillary electrophoresis–mass spectrometry method for small peptides in the soil solution. Soil Biol Biochem 63:80–84
Warren CR (2013b) High diversity of small organic N observed in soil water. Soil Biol Biochem 57:444–450
Warren CR (2013c) Quaternary ammonium compounds can be abundant in some soils and are taken up as intact molecules by plants. New Phytol 198:476–485
Warren CR, Taranto MT (2010) Temporal variation in pools of amino acids, inorganic and microbial N in a temperate grassland soil. Soil Biol Biochem 42:353–359
Watrous J, Roach P, Alexandrov T, Heath BS, Yang JY, Kersten RD, van der Voort M, Pogliano K, Gross H, Raaijmakers JM, Moore BS, Laskin J, Bandeira N, Dorrestein PC (2012) Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci U S A 109:E1743–E1752
Watrous JD, Dorrestein PC (2011) Imaging mass spectrometry in microbiology. Nat Rev Microbiol 9:683–694
Welsh DT (2000) Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. FEMS Microbiol Rev 24:263–290
Werdin-Pfisterer NR, Kielland K, Boone RD (2009) Soil amino acid composition across a boreal forest successional sequence. Soil Biol Biochem 41:1210–1220
Wessel-Bothe S, Pätzold S, Klein C, Behre G, Welp G (2000) Adsorption von Pflanzenschutzmitteln und DOC an Saugkerzen aus Glas und Keramik. J Plant Nutr Soil Sci 163:53–56
Whiffen LK, Midgley DJ, Mcgee PA (2007) Polyphenolic compounds interfere with quantification of protein in soil extracts using the Bradford method. Soil Biol Biochem 39:691–694
Wichern F, Lobe I, Amelung W, Muller T, Joergensen RG, Buerkert A (2004) Changes in amino acid enantiomers and microbial performance in soils from a subtropical mountain oasis in Oman abandoned for different periods. Biol Fertil Soils 39:398–406
Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia JG, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong YP, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I (2009) HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res 37:D603–D610
Wood JM, Bremer E, Csonka LN, Kraemer R, Poolman B, van der Heide T, Smith LT (2001) Osmosensing and osmoregulatory compatible solute accumulation by bacteria. Comp Biochem Physiol Mol Integr Physiol 130:437–460
Yu Z, Zhang Q, Kraus TEC, Dahlgren RA, Anastasio C, Zasoski RJ (2002) Contribution of amino compounds to dissolved organic nitrogen in forest soils. Biogeochemistry 61:173–198
Yu ZS, Kraus TEC, Dahlgren RA, Horwath WR, Zasoski RJ (2003) Mineral and dissolved organic nitrogen dynamics along a soil acidity-fertility gradient. Soil Sci Soc Am J 67:878–888
Zabowski D (1989) Limited release of soluble organics from roots during the centrifugal extraction of soil solutions. Soil Sci Soc Am J 53:977–979
Zhang T, Creek DJ, Barrett MP, Blackburn G, Watson DG (2012) Evaluation of coupling reversed phase, aqueous normal phase, and hydrophilic interaction liquid chromatography with orbitrap mass spectrometry for metabolomic studies of human urine. Anal Chem 84:1994–2001
Acknowledgments
This work was supported by a Future Fellowship from the Australian Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Philippe Hinsinger.
Rights and permissions
About this article
Cite this article
Warren, C.R. Organic N molecules in the soil solution: what is known, what is unknown and the path forwards. Plant Soil 375, 1–19 (2014). https://doi.org/10.1007/s11104-013-1939-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-013-1939-y