
Abstract
Although the human immunodeficiency virus (HIV) causes one of the most important infectious diseases worldwide, attempts to develop an effective vaccine remain elusive. Designing recombinant proteins capable of eliciting significant and protective mammalian immune responses remain a priority. Moreover, large-scale production of proteins of interest at affordable cost remains a challenge for modern biotechnology. In this study, a synthetic gene encoding a C4V3 recombinant protein, known to induce systemic and mucosal immune responses in mammalian systems, has been introduced into tobacco chloroplasts to yield high levels of expression. Integration of the transgene into the tobacco plastome has been verified by Southern blot hybridization. The recombinant C4V3 protein is also detected in tobacco chloroplasts by confocal microscopy. Reactivity of the heterologous protein with both an anti-C4V3 rabbit serum as well as sera from HIV positive patients have been assayed using Western blots. When administered by the oral route in a four-weekly dose immunization scheme, the plant-derived C4V3 has elicited both systemic and mucosal antibody responses in BALB/c mice, as well as CD4+ T cell proliferation responses. These findings support the viability of using plant chloroplasts as biofactories for HIV candidate vaccines, and could serve as important vehicles for the development of a plant-based candidate vaccine against HIV.
Similar content being viewed by others


Introduction
The development of an effective vaccine against the Human Immunodeficiency Virus (HIV)/AIDS has not yet been achieved, and the natural immune response in humans against HIV is incapable of eradicating HIV once primary infection has been established (Johnston and Fauci 2008). To date, only three candidate vaccines have completed phase III clinical efficacy trials, with two poor efficacy results. The latter include the Env gp 120 vaccine–AIDSVAX and the recombinant Adenovirus 5 HIV-1 gag/pol/nef trivalent vaccine-STEP trial (Barouch and Korber 2010). Therefore, the development of a safe, effective, environmentally friendly delivery, and affordable AIDS vaccine formulation is urgently needed. The efficacy of such a HIV vaccine has been associated with the induction of broad neutralizing antibodies and cellular responses (Kuroda et al. 1999; Ferrantelli et al. 2004).
The glycoprotein (gp) 120 subunit is an important component of the HIV viral envelope as it participates in interactions between receptors of the host cell and the virus surface. Thus, humoral responses against this gp120 component may block virus entry. However, a recombinant gp120 has failed to demonstrate efficacy in clinical trials (Russell et al. 2004; Pitisuttithum et al. 2006). Currently, a number of strategies are being pursued to design immunogens that can broadly elicit neutralizing antibodies against gp120 (Pantophlet and Burton 2006).
Thus far, a number of gp120 epitopes have been characterized (Pancera et al. 2010). Among these, the V3 loop is of particular interest as several studies have reported that anti-V3 antibodies present in sera of patients as well as immunized guinea pigs and monkeys are capable of neutralizing a number of primary HIV isolates that in turn can block HIV entry into host cells by interfering with the Env component. It is known that these neutralizing antibodies can prevent HIV infection following intravenous, vaginal, rectal, and oral challenges in non-human primates, thus indicating that V3 can serve as an adequate vaccine target (Liao et al. 2000; Yang et al. 2004; Chakrabarti et al. 2005).
Synthetic peptides comprising the V3 loop and the C4 domain from gp120 have been reported to be immunogenic, and this is probably due to the role of C4 in binding to the CD4 receptor (Graham et al. 2010). Although synthetic peptides have been used in efforts to characterize properties of C4V3 antigens as candidate vaccines, this approach has been limited to clinical trials due to the high cost of synthesis of these peptides. In this context, Varona-Santos et al. (2006) have designed a C4V3 polypeptide based on the MN HIV isolate. This protein, designated as rC4V3, has been produced in Escherichia coli as an expression host, yielding up to 75 mg of C4V3 per liter of culture, and this has been subsequently purified by Immobilized Metal Affinity Chromatography (IMAC). This rC4V3 protein has been found to be immunogenic in BALB/c mice when administered by both intranasal (i.n.) and intramuscular (i.m.) routes and without the use of adjuvants. Therefore, the rC4V3 protein is a promising candidate for pursuing HIV immunization studies. However, scale-up production of rC4V3 using E. coli as an expression system also requires the use of fermentation technology in a well-defined culture medium for production, followed by several purification steps for recombinant protein extraction. Therefore, utilizing alternative large-scale production systems for rC4V3 will overcome some of these features, and would lead to low-cost production of this recombinant protein. One such platform is the use of genetically engineered plants, serving as biofactories for production of plant-based vaccines against a wide variety of human infectious and autoimmune diseases (Paul and Ma 2010; Soria-Guerra et al. 2011). This concept has led to a number of clinical trials with promising results (Yusibov et al. 2011). Plant-based vaccines can be produced at low-cost, are easy to store, are free of mammalian pathogens when grown under appropriate conditions, as well as free of bacterial products that can function as Pathogen-Associated Molecular Patterns (PAMPs). In addition, oral administration of edible plant tissues may induce mucosal immune responses allowing for production of local antibodies that could block pathogen entry before primary infection takes place at the lamina propria (Meyers et al. 2008). This approach will also avoid all purification steps, thus drastically reducing cost of production. Although not edible, certain plants such as tobacco and Arabidopsis are easily genetic engineered in a short period of time, and therefore these species have been initially used to provide a ‘proof the concept’ of plant-based oral immunization (Lau et al. 2010; Gonzalez-Rabade et al. 2011).
Transplastomic approaches have been used as suitable strategies to obtain high levels of accumulation of recombinant proteins. Immunogens such as the cholera toxin, LTB-ST fusion protein from E. coli, and the F-V fusion protein from Yersinia pestis, among others, have been expressed in plants, and their immunogenicity have been confirmed in test animal models (reviewed by Lössl and Waheed 2011; Soria-Guerra et al. 2011).
Herein, we describe the development of transplastomic tobacco plants expressing a C4V3 polypeptide, which was found to be immunogenic as it induced both mucosal and systemic antibody responses in test mice.
Materials and methods
Construction of the expression vector
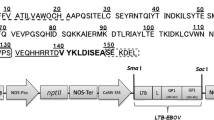
The C4V3 encoding gene has been previously described by Varona-Santos et al. (2006). This gene comprises a V3 loop from the MN isolate as well as a C4 peptide fused to the N terminus of V3 along with a His tag at the carboxi terminus. The C4V3 gene has been modified by PCR using primers to add XbaI and XhoI restriction sites at the 5′ and 3′ ends, respectively (underlined nucleotides), to facilitate cloning; moreover, a ribosome binding site upstream of the start codon (nucleotides in italics) has been also added (Fig. 1a). The following primer sequences have been used: Forward, 5′GGCTATCTAGAAGGGAGGGACATATGGATCTGGAAAAACAAAT; Reverse, 5′CCATAGCTCGAGTCAATGATGATGATGATGATG.

a Physical map of the transformation vector pBic-C4V3. A derivative of the pBic vector was constructed by cloning the C4V3 gene downstream of the Prrn promoter. This vector targets the integration of the expression cassette into the inverted repeat region of the chloroplast genome. Arrows indicate landing sites for oligonucleotides 1, 2, and 3. The black box represents the chloroplast genome close to the insertion site. The black bar represents the probe used for Southern blot hybridization. b PCR analysis showing C4V3 transgene integration into the chloroplast genome of tobacco. PCR was performed with a primer set comprised of oligonucleotide 3 that lands on the tobacco plastome adjacent to INSL, and oligonucleotide 2 that lands on the C4V3 gene. Presence of amplicons of ~1,800 bp indicates the insertion of the C4V3 transgene at the site specific integration. Lane: 1, 1 kb ladder; lanes 2–5, candidate lines NR3, NR4, NR7, NR8; and lane 6, wild-type plant
The amplicon was digested with XbaI and XhoI enzymes and cloned into the pBic vector downstream of the Prrn promoter, which has been constructed as described previously by Rosales-Mendoza et al. (2009). This vector would allow for expression of the C4V3 gene as a bicistron along with the aadA gene, a selectable marker gene conferring spectinomycin resistance to transformed cells. The final sequence of the pBic-C4V3 vector was subsequently verified by enzyme profiling and by sequencing.
Transplastomic tobacco development
Plant transformation was conducted according to the protocol described by Daniell et al. (2005). Briefly, surface-sterilized seeds of tobacco, Nicotiana tabacum cv. Petite Havana SR1, were germinated on Murashige and Skoog (MS) medium. Fully-expanded leaves from 1 month-old seedlings were excised, placed with the abaxial side up in plates containing the RMOP medium, and bombarded with DNA-coated gold particles at 1,100 psi pressure using the PDS-1000/He biolistic microprojectile gene gun (Bio-Rad, Hercules, CA). Following bombardment, leaf tissues were maintained in the dark for 48 h, then cut into 50 mm2 segments, and incubated on a selection medium consisting of RMOP medium and containing 500 mg/l spectinomycin. Explants were then transferred to fresh medium once every 2 weeks, and divided into 50 mm2 until shoots developed.
Leaf segments from spectinomycin-resistant shoots were used to conduct the second round of selection. Finally, a third round of selection was done using leaf sections from shoots obtained in the second round of selection. After the third round of selection, regenerated shoots were rooted on a plant growth regulator (PGR)-free MS medium containing spectinomycin. When roots were 2 cm in length, plantlets were transferred to pots containing Sunshine soil mix. Plants were grown at 24°C under a 16 h photoperiod (100 μmol m−2s−1).
Detection of transformation events
Total DNA was isolated from leaves of both putative transformants and wild-type plants according to Dellaporta et al. (1983). PCR conditions were performed as follows: A 50 μl reaction mixture containing 100 ng DNA, 1.5 mM magnesium chloride, 2.5 U Taq DNA polymerase (Vivantis Technologies, Selangor, Malaysia), 1 mM dNTPs, and 1 μM of each of forward and reverse primers. Temperature cycling conditions were as follows: 94°C for 2 min (initial denaturation); 35 cycles of 95°C for 30 s (denaturation); 58°C for 30 s (annealing); 72°C for 2 min (elongation); and a final extension at 72°C for 5 min. PCR products were analyzed by electrophoresis on 1% agarose gels. Primers used for PCR analysis included the following: primer 1 (5′ggatccatggtgaaagttaaatgttacgttctttt) and primer 2 (5′cagtcgcaattgaattaggagtcttgttgttcc), amplifying the C4V3 sequence, as well as primer 3 (5′aagaatgggtgagggtattctgcctaaata) that lands on the native chloroplast genome. The latter was included to confirm site specific integration of the expression cassette into the tobacco plastome by double homologous recombination.
For Southern blot analysis, total DNA (20 μg) was digested with HindIII, electrophoresed on a 1.0% agarose gel, and transferred to a nylon membrane (Amersham BioSciences, Piscataway, NJ). Then, DNA was cross-linked using a transilluminator, and the blot was prehybridized with a DIG Easy Hyb (Roche, Indianapolis, IN). A C4V3 DIG-labelled probe was developed using the PCR-DIG Probe Synthesis Kit (Roche Co., Mannheim, Germany) with pBic-C4V3 vector used as a template. Following hybridization at 42°C for 16 h, the membrane was washed in an SSC buffer series (2–0.5×), and a gene-specific signal was detected using the DIG Detection Kit following the manufacturer’s instructions (Roche Co., Mannheim, Germany).
Rabbit immunization with rC4V3
A 4-month-old New Zealand White rabbit (3 kg average weight) was used to produce a hyperimmune C4V3 anti-serum for immundetection of the tobacco-derived C4V3. For immunization, a pure antigen (400 μg) was produced as described by Varona-Santos et al. (2006), and administered by the subcutaneous (sc) route in six doses. The rabbit was primed with a dose of 400 μg of rC4V3 in 200 μl of PBS plus 200 μl of complete Freund’s adjuvant, and then subjected to 5 weekly doses consisting of 400 μg of rC4V3 plus 200 μl of incomplete Freund’s adjuvant. Blood collection was performed at week 7 by a puncture to the ear vein. Blood samples were incubated at 25°C for 1 h to facilitate clotting, and then centrifuged at 5,000 rpm for 5 min at 4°C. The anti-serum was collected, and immunoreactivity was assessed by both Western blot and ELISA using plates coated with pure rC4V3 (Varona-Santos et al. 2006).
Detection of tobacco-derived C4V3
Microscopy—Tobacco leaves were fixed in a freshly prepared ZSF solution consisting of 0.1 M Tris base buffer with 0.05% calcium acetate [pH 7–7.4], and containing 0.5% zinc acetate along with 0.5% zinc chloride. To allow penetration of the fixative, leaf tissues were vacuum-infiltrated. Leaf sections were dehydrated in a gradient series of ethyl alcohol, and imbedded in paraffin.
An indirect immunofluorescence technique was used to detect C4V3 on 5 μm microtome-cut mounted leaf sections. Non-specific binding was blocked with 5% BSA/0.05% Triton× 100 in 0.01 M PBS, pH 7.0, for 30 min. Sections were incubated with anti-C4V3 rabbit serum (1:1,000 dilution in PBS plus 0.07% Triton X-100), followed by incubation with a secondary antibody, goat anti-rabbit FITC (1:1,000 dilution in PBS plus 0.07% Triton X-100; Invitrogen, USA). Samples were dehydrated and covered with Entellan (Merck, Darmstadt, Germany). Control experiments for staining specificity included either using appropriate isotype-matched non-specific antibodies or replacement of primary antibodies with non-immune serum and omission of primary antibody from incubation protocols. No specific staining was detected in either case. Immunofluorescence images were captured using a confocal laser scanning microscope (TCS SP2; Leica Microsystems AG, Wetzlar, Germany).
ELISA—About 100 mg of fresh leaf tissue was grounded, and resuspended in 500 μl protein extraction buffer (100 mM NaH2PO4, 8 M Urea, and 0.5 M NaCl; pH 8). Samples were centrifuged at 14,000 rpm in a microcentrifuge for 10 min at 4°C. Assay plates were coated overnight at 4°C with protein extracts diluted in carbonate buffer. Plates were washed with PBST, and blocked with 2% fat- free dry milk for 1 h at room temperature. After washing plates with PBST, anti-C4V3 anti-sera (1:500) were added for 1 h. A horseradish peroxidase-conjugated anti-rabbit IgG (1:10,000 dilution; Sigma, Missouri, USA) was added and incubated for 1 h. After washing with PBST, a substrate solution composed of 0.3 mg/l 2-2′-Azino-bis-3 etilbenztiasoline-6-sulphuric acid (ABTS; Sigma, Missouri, USA) and 0.1 M citric acid, pH 4.35, was added. The OD at 405 nm was recorded in a Multiskan Ascent (Thermo Scientific, Massachusetts, USA) microplate reader. A pure recombinant C4V3 (1–10 μg) was used as a standard to calculate C4V3 levels, expressed as μg of C4V3 per g fresh weight tobacco.
Western blot- Total soluble proteins were extracted by resuspending 10 mg of freeze-dried leaf tissue into 50 μl of 1× reducing loading buffer. Samples were denatured by boiling for 5 min at 95°C, and debris was eliminated by centrifugation at 12,000×g for 10 min. The supernatant was subjected to SDS-PAGE in 10% acrylamide gels. The gel was blotted onto a BioTrace PVDF membrane (Pall Corporation, NY). After blocking in PBST plus 5% non-fat milk, blots were incubated with primary antibodies directed against either the His tag (1:2,000 dilution; GenScript Co., New Jersey, USA) or C4V3 (1:2,000 dilution). A pool of sera from HIV-positive patients was also used for primary labelling. A secondary horseradish peroxidase conjugated anti-human, anti-rabbit, or anti-mouse antibody (1:10,000 dilution; Sigma, Missouri, USA) was added for 2 h at room temperature. Antibody binding was detected by incubation with a SuperSignal West Dura solution following the manufacturer’s instructions (Thermo Scientific, Massachusetts, USA). Signal detection was verified using an X-ray film following standard procedures.
Mice immunization
Immunogenicity of tobacco-derived C4V3 was evaluated in female BALB/c mice (8–11 weeks of age) that were housed in filter-topped cages, and cared for according with federal regulations for animal experiments (NOM-062-ZOO-1999, Ministry of Agriculture, Mexico). The immunization protocol was approved by the Institutional Animal Care and Use Committee (IACUC).
Mice were randomly divided into three different groups (n = 5) receiving the following treatments: 50 mg of freeze-dried tobacco leaf powder from transgenic line NR7 containing approximately 15 μg of C4V3 (group TT), 150 μg of E. coli-derived C4V3 (group+), 50 mg of freeze-dried tobacco leaf powder from wild-type plants (group WT), and the vehicle alone (group PBS). Four weekly doses were administered by oral feeding to each group on days 0, 7, 14, and 21. Mice were sacrificed on day 28, and serum samples were collected from blood extracted following cardiac puncture.
To detect mucosal antibodies, intestines were collected, and 5 ml of cold RPMI medium was flushed through. This suspension was mixed, and 500 μl of 10 mM p-hydroxymercuribenzoate, dissolved in 150 mM Tris-base, was added. Samples were centrifuged at 12,000×g at 4°C for 10 min, and recovered supernatants were stored at −70°C until used for analysis.
ELISA assays
The anti-C4V3 antibody content was determined by enzyme-linked immunosorbent assay (ELISA). For each reagent/sample used in this assay, 100 μl were used, unless noted otherwise. Plates were coated with a pure C4V3 protein (10 μg/ml) diluted in a carbonate/bicarbonate buffer (15 mM Na2CO3 and 35 mM NaHCO3; pH 9.6), incubated overnight at 4°C, and blocked with 5% fat-free milk dissolved in PBS (100 mM NaCl, 10 mM Na2HPO4, and 3 mM KH2PO4; pH 7.2). Serum samples (dilution 1:40) or intestinal samples (dilution 1:2), diluted in PBST (0.05% v/v Tween-20 in PBS), were added. Triplicates from each sample were incubated overnight at 4°C. Either a horseradish peroxidase-conjugated goat anti-mouse anti-immunoglobulin G (IgG) (Pierce, Illinois, USA) or anti-IgA (Zymed, California, USA) was then added to each well, and plates were incubated for 1 h at 37°C. Plates were washed, and the ABTS substrate solution was added. The OD at 405 nm was recorded in a Multiskan Ascent (Thermo Scientific, Massachusetts, USA) microplate reader.
T-cell proliferation and IFN-γ production assays
Spleens from mice groups were removed aseptically, and used to establish single cell suspension cultures. Cells were washed in 10 ml of RPMI 1640, and then resuspended in R-10 consisting of RPMI medium supplemented with 10% of fetalbovine serum (GIBCO, California, USA), 2 mM l-glutamine, 10 mM Hepes, 1 mM sodium piruvate, and 1% vol/vol non-essential amino acid solution (Sigma, Missouri, USA). Cell viability was evaluated using a Trypan Blue exclusion dye, while cell density was estimated using a Neubauer chamber, and adjusted to 2 × 106 cells/ml with culture medium. Two splenocyte pools were obtained from each experimental group, comprised of cells from two different animals, and these were used for both proliferation and IFN-γ production assays.
To perform proliferations assays, freshly prepared splenocytes were stained with CFSE (5,6-carboxyfluorescein diacetate succinimidyl ester) as previously described (Saavedra et al. 2001). CFSE-stained splenocytes (1 × 106 cells) were cultured in 1 ml of culture medium with or without pure C4V3 (1 μg/ml), in triplicates, for 65 h at 37°C in 5% CO2. Cells were harvested, washed with PBS-1% FBS-0.1% NaN3, and stained for 30 min (4°C) with either phycoerythrin (PE)-labelled anti-CD4 monoclonal antibody (clone RM4-5, Caltag, CA, California, USA) or with PE-labelled anti-CD8 monoclonal antibody (clone 5H10-1) (BD, California, USA). Cells were then washed three times with the same buffer, resuspended in PBS, and analyzed by flow cytometry.
To estimate IFN-γ production, splenocytes were cultured on RPMI 1640 medium (GIBCO BRL, California, USA) supplemented with 10% heat- inactivated fetal bovine serum (FSB), 100 U/ml penicillin G, and 100 μg/ml streptomycin. Cell cultures were established in either presence or absence of C4V3 pure protein (μg/ml) at 37°C in a humid environment with 5% CO2 for 1 h. Then, cells were incubated with 10 μg/ml of brefeldin A (Sigma, Missouri, USA) for 4 h under the same conditions. Stimulated cells were washed in ice-cold phosphate-buffered saline solution containing 3% FBS and 0.05% sodium azide. Approximately 1 × 106 cells were labelled with 0.1 μg of each of phycoerythrin (PE) labelled with anti-CD4 (Caltag, California, USA), PE-Cy5 labelled anti-CD3 (eBioscience, California, USA), APC labelled anti-CD8 mouse antibodies (Biolegend, California, USA), and isotype matched controls (BD, California, USA) in a total volume of 100 μl of staining buffer for 30 min. Subsequently, cells were permeabilized with citofix-citoperm for 15 min and labelled with 0.1 μg of fluorescein isothiocyanate (FITC) labelled anti-IFN-γ (BD, California, USA). These cells were washed with a cold staining buffer containing 0.05% sodium azide.
Flow cytometry analysis was performed using a FACSCalibur flow cytometer (BD, California, USA). Lymphocytes were gated according to forward and side scattering, and at least 250,000 events were acquired and analyzed. Frequency of positive events was calculated against relevant staining controls.
Data analysis
Statistical significance (P-values) was determined using one-way ANOVA. Statistical analysis was performed with Statistica software (version 2.7), and student tests for independent samples at P = 0.05 were conducted.
Results
Transplastomic tobacco plants carrying the C4V3 transgene
In this study, the pBic vector was selected to drive the transfer and expression of the C4V3 gene. The strategy based on the use of site-directed mutagenesis and restriction with XbaI and XhoI allowed for successful cloning of the gene into the pBic vector, yielding the pBic-C4V3, as confirmed by sequencing. A positive clone was propagated, and used for biolistic bombardment. This vector is schematically represented in Fig. 1a.
Following bombardment, leaf explants were successfully maintained under aseptic conditions. The majority of explants turned white in coloration 3 weeks following bombardment. Callus development was observed at 6 weeks following incubation on the selection medium. A total of 10 putative transformed lines were induced on the selection medium, and these were subjected to regeneration/selection rounds as described above. Six months following bombardment, regenerated shoots from four lines were successfully rooted, transferred to soil, and grown in the greenhouse. Putative transgenic lines were designated as NR3, NR4, NR7, and NR8, and used for further analysis. When compared to wild-type plants (control), plants from putative transgenic lines NR3, NR7, and NR8 exhibited some phenotypic modifications, characterized by slight chlorosis and lack of seed development.
Detection of the C4V3 transgene in tobacco plants
Presence of the C4V3 transgene was assessed by PCR analysis. When C4V3 primers were used, 250 bp amplicons were detected in all four analyzed lines, thus confirming presence of the transgene (data not shown). Similarly, PCR analysis using a primer landing on the chloroplast genome outside of the recombination region along with a primer landing on the C4V3 transgene identified the expected 1,800 bp amplicon in transplastomic lines. No amplicons were detected from wild-type tobacco plants (Fig. 1b). These analyses confirmed successful site-directed introduction of the C4V3 transgene into the tobacco plastome.
For Southern blot analysis, total DNA samples were digested with HindIII, a restriction site within the expression cassette, downstream of the aadA gene and left of the recombination flanking sequences (see Fig. 1a). Hybridization with a C4V3 specific probe revealed that all putative transplastomic lines showed the expected 1,400 bp band, corresponding to the fragment yielded following HindIII digestion. No chemilluminicent signal was detected in DNA samples from the wild-type tobacco plant (Fig. 2).

Detection of C4V3 insertion by Southern blot analysis. Plants from transplastomic lines NR3, NR4, and NR7 were analyzed by digesting DNA samples with HindIII and detecting C4V3 sequences by hybridization. Lanes 1–3, T0 plants from lines NR3, NR4, and NR7; lanes 4–6, plants from line NR7; lane 7, WT plant; lane 8, DIG-labelled molecular weight marker. The single 1,400 bp band showed presence of the inserted C4V3 cassette
Moreover, seeds of transplastomic line NR7 were capable of germinating on a selection medium containing spectinomycin; while, none of the WT seeds germinated when grown on this selection medium (Fig. 3).

Spectinomycin-based selection of tobacco seedlings. Seeds from either the transplastomic NR7 line (NR7) or wild-type (WT) plant were germinated on MS medium containing 500 mg/l spectinomycin. Note the chlorosis developed by WT seedlings; while, seedlings of NR7 line were green and capable of growing on the selection medium
Localization and antigenicity of tobacco-derived C4V3
To demonstrate that the C4V3 protein was specifically produced in chloroplasts of transplastomic plants, leaves from the different transplastomic lines were subjected to an immunohistofluorescence assay using the anti-C4V3 serum, followed by staining with an FITC labelled secondary antibody. As a marker for detecting chloroplasts, tobacco leaves were analyzed for chlorophyll fluorescence using confocal microscopy (Ex 488 nm, Em 687 nm), as it is well established that chlorophyll, a major contributor to endogenous auto-fluorescence, has an absorption band in the blue region of the visible spectrum that produces a significant amount of fluorescence at wavelengths >600 nm when excited with a wavelength of 488 nm. Confocal microscopy revealed strong FITC signals (Ex 488 nm, Em 520 nm) that co-localized with the chlorophyll autofluorescence. These findings indicated that expression of the C4V3 transgene must be located within chloroplasts as a result of the regulatory sequences contained in the expression cassette and the site-directed insertion in the chloroplast genome. No significant FITC signal was detected in leaves from WT plants (Fig. 4).

Detection of the C4V3 protein in tobacco chloroplasts. Tobacco leaves from either wild type (WT) or transplastomic tobacco (NR-7) were labelled with rabbit anti-C4V3 serum, and then stained with FITC conjugated secondary antibody. Laser confocal images show chloroplasts accumulating C4V3 (green signal). Chlorophyll autofluorescence was also captured in order to localize chloroplasts (blue signal). The overlap of the two spectra is shown in the right-hand set of photographs. Bars 47.57 or 13.61 μm
To further confirm expression of the immunoreactive C4V3 protein, ELISA assays were performed with denatured protein extracts. Significant OD signals were recorded for transplastomic lines when compared to those observed for the WT plant. Accumulation levels estimated based on the standard curve were up to 25 μg/g fresh weight (FW) tobacco leaves.
Western blot assays against C4V3 components were also carried out. Significant signals were detected when primary labelling was performed with the different antibodies, including anti-His, anti-C4V3, and sera from HIV-positive patients (Fig. 5a–c). However, the molecular weight of the immunoreactive protein was higher than expected, as previously noted by Varona-Santos et al. (2006). These results suggested that tobacco chloroplasts were capable of expressing the C4V3 and retaining its antigenic determinants.

Western blot analyses for the detection of the tobacco derived C4V3. Total soluble protein extracts were resolved by SDS PAGE and blotted for the immunodetection by a anti-His antibody; b C4V3 anti-serum; c sera from HIV-positive patients; and d sera from HIV-negative patients. For a and b, lanes correspond to the following: 1, 250 ng of pure purified C4V3; 2, 500 ng of pure C4V3; 3 and 4, WT plants; and 5–7, lines NR3, NR4 and, NR7. For c, lanes correspond to the following: 1, 250 ng of pure purified C4V3; 2, 500 ng of pure C4V3; 3 and 4, WT plants; 5 and 6, lines NR3, and NR7. For d, lanes correspond to the following: 1–4, lines NR3, NR4, NR7, and NR8; 5 and 6, WT plants; and 7, 500 ng of pure C4V3
Tobacco-derived C4V3 is immunogenic in mice
Leaves from adult plants of transplastomic line NR7 along with WT plants were freeze-dried for 24 h, and then ground into a fine powder to pursue immunization assays. To assess the oral immunogenicity of the tobacco-based C4V3 antigen, BALB/c mice were immunized using four oral doses of either the vehicle alone (PBS) or a soluble protein extracted from either freeze-dried tobacco leaves from either line NR7 (TT) or WT. An additional group was immunized with E. coli-derived C4V3 to assess oral immunogenicity of C4V3. After collecting sera and intestinal samples from test mice, antibody contents were evaluated by ELISA.
Interestingly, E. coli-derived C4V3 was found to be immunogenic when administered orally. Anti-C4V3 serum antibody levels in control mice immunized with WT tobacco were not different from those of mice treated with the PBS vehicle alone; whereas, mice immunized with transplastomic tobacco expressing C4V3 elicited significant IgG serum anti-C4V3 responses (P < 0.05) (Fig. 6a). Similar results were obtained from intestinal fluids wherein significant anti-C4V3 responses were observed for both serum IgG and mucosal IgA (Fig. 6b). No significant differences were found between groups immunized with E. coli-derived C4V3 and those immunized with plant-derived C4V3. These findings demonstrated that chloroplast-derived C4V3 was immunogenic via oral delivery.

Anti-C4V3 immune responses elicited by oral immunization of BALB/c female mice with tobacco derived C4V3 (TT) or E. coli derived C4V3 (+). Animals were immunized orally using a 4 weekly-dose scheme, and levels of IgG in sera diluted 1:40 (a) and IgA in intestinal fluids diluted 1:2 (b) were measured by ELISA. Comparisons showing significant statistical differences among treatments, made against the WT and PBS groups, are indicated by an asterisk (P < 0.05)
To assess the magnitude of the rC4V3-specific response induced by TT leaf tissue in test mice, both T cell proliferation and production of IFN-γ, following stimulation for 65 h with either rC4V3 or with medium alone, were determined. For the T-cell proliferation, a significant increase from 0.51% to 3.15% of CD4+ proliferation responses was observed in mice immunized with TT leaf tissue; whereas, proliferating responses of mice treated with WT tobacco showed values ranging between 1.06 and 1.26% (Fig. 7). Only a slight increase in the frequency of IFN-γ producing CD8+ T cells was observed when these were stimulated with rC4V3 (data not shown). No proliferating responses were observed for cells from non-immunized mice whether or not they were peptide-stimulated (data not shown). Significantly higher frequencies of IFN-γ-producing CD4+ T cells were detected in C4V3-stimulated splenocyte pools of mice immunized with TT leaf tissue (2.77%) in comparison with those treated with WT tobacco (0.97%) (Fig. 8). Therefore, these findings provided further evidence of the cellular immune response elicited by oral feeding of mice with tobacco-derived C4V3.

CD4+ T cells proliferation assays. Pools of splenocytes from mice immunized with either tobacco-derived C4V3 (TT) or wild type tobacco (WT) were cultured in the presence or absence of pure C4V3. Proliferation was detected by CSFE staining, and antibody labelling followed by flow cytometer analysis. A representative result obtained from a pool of cells is presented. Note the significant increase in CD4+ T cell proliferation in the TT group when cultured in the presence of C4V3

IFN-γ production by CD4+ T cells stimulated with C4V3. Pools of splenocytes from mice immunized with either tobacco-derived C4V3 (TT) or wild type tobacco (WT) were cultured in the presence or absence of pure C4V3. IFN-γ producing CD4+ T cells were detected by staining with labelled antibodies followed by flow cytometer analysis. A representative result obtained from a pool of cells is presented. Note the significant increase of IFN-γ producing CD4+ T cells in the TT group when cultured in the presence of C4V3
Discussion
Previously, we have reported on the production of the C4V3 protein at high levels in recombinant E. coli, demonstrating that despite its low molecular weight, the recombinant C4V3 could induce strong humoral immune responses against HIV-1 sequences when delivered by systemic and mucosal routes, even without the aid of adjuvants (Varona-Santos et al. 2006). In this study, we have engineered tobacco plants to produce a plastid-expressed C4V3 protein. Following particle bombardment and spectinomycin-based selection, four representative transplastomic lines have been recovered and analyzed. Most lines have exhibited slight chlorosis, but only a single line is found to be fertile and has produced seeds (data not shown). Interestingly, previous studies have reported on morphological changes observed in transplastomic plants expressing various HIV antigens (Cueno et al. 2010). Future studies would be conducted to further investigate phenotypic variations observed in these transplastomic lines. Nevertheless, analysis of progeny of transplastomic line NR7, obtained in this study, has been confirmed to be stably transformed and expressing the C4V3 antigen following Southern blot and Western blot analysis, respectively. Based on ELISA and Western blot analyses, accumulation of the recombinant protein reached up to ~25 μg per g FW.
The antigenicity of the recombinant tobacco-based C4V3 was evaluated using a rabbit hyperimmune anti-serum along with sera from HIV-positive patients, confirming the proper display of antigenic determinants of this plant-derived C4V3. However, Western blots revealed signals corresponding to unexpected higher molecular weights, which might be attributed to either polymerization of the C4V3 or formation of complexes with plant endogenous proteins incapable of dissociating even under reducing conditions. It is important to mention that the C4V3 peptide tends to form aggregates following IMAC purification when produced in E. coli (Varona-Santos et al. 2006).
It has been previously reported that a four-dose immunization scheme allows for induction of significant anti-C4V3 immune responses in mice (Staats et al. 1996; Varona-Santos et al. 2006). Thus, the immunogenicity of the tobacco-derived C4V3 was determined following oral dosing of test mice with 4 weekly doses of ~15 μg of plant-derived C4V3. Interestingly, expression levels were high enough to immunize mice with low quantities of freeze-dried tobacco tissue as 50 mg of this tissue contained ~15 μg of C4V3. This dosage minimized any potential toxic effects on test mice.
Detected humoral responses elicited in mice, both at mucosal and systemic levels, have demonstrated that C4V3 produced in tobacco is immunogenic, and therefore could be used for elicitating neutralizing antibodies in future studies. However, this response is lower than that previously observed in mice immunized with E. coli-derived C4V3 via intramuscular and intranasal routes (Esquivel-Pérez and Moreno-Fierros 2005). These observed differences are not unexpected as oral immunization typically triggers only modest responses. Thus, higher doses of the plant-derived antigen are recommended for oral immunization to achieve higher humoral responses.
Previously, we have evaluated two different doses, 10 and 100 μg, of a C4V3 peptide administered to mice by either intramuscular or intranasal routes, along with or without CT (when testing via the intranasal route). Significant and high immune responses have been detected with a 100 μg dose, but only modest responses have been induced with a lower dose (10 μg) co-administered with CT (Esquivel-Pérez and Moreno-Fierros 2005). By administering a much lower dose of an unpurified antigen (15 μg) in this study, the observed immune responses are in fact quite remarkable as they are comparable to those obtained by using 150 μg of E. coli-derived C4V3. It is hypothesized that protective effects against antigen degradation exerted by the matrix constituted by plant cell walls may be responsible for these findings. On the other hand, presence of tobacco-derived C4V3 multimers, detected in western blots, suggests differential aggregation in comparison to that observed with the E. coli derived C4V3 protein. Precipitation in chloroplasts as well as presence of other endogenous proteins in addition to those corresponding to C4V3 may also account for enhanced immunogenicity detected with the plant derived-C4V3. However, additional studies ought to be performed to further clarify these observed findings.
Proliferation and IFN-γ-production evaluated in CD4+ T cells have indicated that CD4+ T cell responses are successfully induced following this immunization protocol. Interestingly, these responses are similar to those induced in previous studies with higher doses (100 μg) of E. coli-derived C4V3 by i.m. and i.n. routes. Therefore, this finding suggests increased immunogenicity of tobacco-derived C4V3 in comparison to that produced in E. coli. As for CD8+ cellular responses, only a slight increase is observed in the TT treated group, which is similar to findings using E. coli-derived C4V3 (Varona-Santos et al. 2006).
In light of results obtained in this study, it is important to note that vaccine costs remain critical for any successful vaccination platform against mammalian pathogens, including HIV, particularly for the vast majority of populations living in developing countries. Plants serve as cost-effective expression systems for large-scale production of vaccine components (Paul and Ma 2010). The use of plant-based vaccines, capable of priming the immune system and resulting in both humoral and mucosal responses directed against infectious agents, has been reported for various pathogens, and particularly for those pathogens that primarily infect mucosal surfaces (Rosales-Mendoza et al. 2009). This approach is also appropriate for HIV as mucosal tissues serve as the major routes for virus entry (Yu and Vajdy 2010). Therefore, antibodies directed against specific HIV antigens could block transcytosis of the virus into the epithelium, thus preventing infection of CD4+ T cells. Interestingly, humoral immune responses have been induced by the transplastomic C4V3 producing line developed in this study, when administered via the oral route.
The relevance of using plants as biofactories for HIV antigens has focused on several components of HIV sequences, including gp120, gp41, tat, p24, and Nef that have been expressed in different food and non-food plants used as production platforms (reviewed by Scotti et al. 2010; Webster et al. 2005). To date, most studies have focused on early HIV antigens produced in plant chloroplasts (Meyers et al. 2008; Scotti et al. 2009). Zhou et al. (2008) have introduced the capsid protein p24 fused to Nef (p24-Nef) into tobacco chloroplasts, and have reported high accumulation of this recombinant fusion protein (40% TSP) in transplastomic lines. More recently, Gonzalez-Rabade et al. (2011) have investigated the immunogenicity of both p24 and p24-Nef proteins, expressed in tobacco chloroplasts, and have found that both recombinant proteins are immunogenic when administered by the s.c. route. Oral immunization with chloroplast derived p24-Nef was capable of enhancing the immune response when used as a boost following s.c. priming.
However, studies on HIV envelope antigens are limited as there are only a few reports on the production of Env epitopes in plants. For example, the pentameric cholera toxin B (CTB) subunit protein has been fused to the V3 loop from gp120, and stably expressed in transgenic potato plants (Kim et al. 2004). Although the CTB-gp120 has been assembled into pentamers and has retained antigenic properties of both components, there is no report on its immunogenicity (Kim et al. 2004). In another report, Matoba et al. (2004) has described the production of a fusion protein comprising CTB and the P1 peptide from gp41 (aa 649–684) in transiently transformed Nicotiana benthamiana plants and in E. coli. The E. coli-derived CTB-P1 has triggered transcytosis-neutralizing serum IgG and mucosal IgA responses when administered to mice orally, thus suggesting that plant-derived CTB-P1 could serve as a mucosal immunogen against HIV.
Taken together, findings presented in this report have further proved the concept that plant chloroplasts are capable of synthesizing a functional C4V3 protein, and this will have implications on the design of immunization strategies against HIV targeting the viral envelop. As tobacco leaves contain significant amounts of phenolics and toxic alkaloids, such as nicotine, the next steps in this research will involve expression of this polypeptide in edible plants, such as lettuce, in order to carry out more detailed immunization experiments with these plant tissues. Moreover, the C4V3 could serve as a foundation for developing novel multiepitopic proteins, including tandem repeats of the same epitope from different isolates. Ongoing studies in our group are pursuing this approach by including repeats of the V3 loop in the same protein.
References
Barouch DH, Korber B (2010) HIV-1 vaccine development after STEP. Annu Rev Med 61:153–167
Chakrabarti BK, Ling X, Yang ZY, Montefiori DC, Panet A, Kong WP, Welcher B, Louder MK, Mascola JR, Nabel GJ (2005) Expanded breadth of virus neutralization after immunization with a multiclade envelope HIV vaccine candidate. Vaccine 23:3434–3445
Cueno ME, Hibi Y, Imai K, Laurena AC, Okamoto T (2010) Impaired plant growth and development caused by human immunodeficiency virus type 1 Tat. Transgenic Res 19:903–913
Daniell H, Ruiz ON, Dhingra A (2005) Chloroplast genetic engineering to improve agronomic traits. Methods Mol Biol 286:111–138
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Esquivel-Pérez R, Moreno-Fierros L (2005) Mucosal and systemic adjuvant effects of cholera toxin and Cry1Ac protoxin on the specific antibody response to HIV-1 C4/V3 peptides are different and depend on the antigen co-administered. Viral Immunol 18:695–708
Ferrantelli F, Rasmussen RA, Buckley KA, Li PL, Wang T, Montefiori DC, Katinger H, Stiegler G, Anderson DC, McClure HM, Ruprecht RM (2004) Complete protection of neonatal macaques against oral challenge with pathogenic simian-human immunodeficiency virus by human anti-HIV monoclonal antibodies. J Infect Dis 189:2167–2173
Gonzalez-Rabade N, McGowan EG, Zhou F, McCabe MS, Bock R, Dix PJ, Gray JC, Ma JK (2011) Immunogenicity of chloroplast-derived HIV-1 p24 and a p24-Nef fusion protein following subcutaneous and oral administration in mice. Plant Biotechnol J 9:629–638
Graham BS, McElrath MJ, Keefer MC, Rybczyk K, Berger D, Weinhold KJ, Ottinger J, Ferarri G, Montefiori DC, Stablein D, Smith C, Ginsberg R, Eldridge J, Duerr A, Fast P, Haynes BF (2010) Immunization with cocktail of HIV-derived peptides in Montanide ISA-51 is immunogenic, but causes sterile abscesses and unacceptable reactogenicity. PLoS ONE 5:e11995
Johnston MI, Fauci AS (2008) An HIV vaccine-challenges and prospects. N Engl J Med 359:888–890
Kim TG, Gruber A, Langridge WH (2004) HIV-1 gp120 V3 cholera toxin B subunit fusion gene expression in transgenic potato. Protein Expr Purif 37:196–202
Kuroda MJ, Schmitz JE, Charini WA, Nickerson CE, Lifton MA, Lord CI, Forman MA, Letvin NL (1999) Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. J Immunol 162:5127–5133
Lau OS, Ng DW, Chan WW, Chang SP, Sun SS (2010) Production of the 42-kDa fragment of Plasmodium falciparum merozoite surface protein 1, a leading malaria vaccine antigen, in Arabidopsis thaliana seeds. Plant Biotechnol J 8:994–1004
Liao HX, Etemad-Moghadam B, Montefiori DC, Sun Y, Sodroski J, Scearce RM, Doms RW, Thomasch JR, Robinson S, Letvin NL, Haynes BF (2000) Induction of antibodies in guinea pigs and rhesus monkeys against the human immunodeficiency virus type 1 envelope: neutralization of nonpathogenic and pathogenic primary isolate simian/human immunodeficiency virus strains. J Virol 74:254–263
Lössl AG, Waheed MT (2011) Chloroplast-derived vaccines against human diseases: achievements, challenges and scopes. Plant Biotechnol J 9:527–539
Matoba N, Magérus A, Geyer BC, Zhang Y, Muralidharan M, Alfsen A, Arntzen CJ, Bomsel M, Mor TS (2004) A mucosally targeted subunit vaccine candidate eliciting HIV-1 transcytosis-blocking Abs. Proc Natl Acad Sci USA 101:13584–13589
Meyers A, Chakauya E, Shephard E, Tanzer FL, Maclean J, Lynch A, Williamson AL, Rybicki EP (2008) Expression of HIV-1 antigens in plants as potential subunit vaccines. BMC Biotechnol 8:53
Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD (2010) Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA 107:1166–1171
Pantophlet R, Burton DR (2006) GP120: target for neutralizing HIV-1 antibodies. Annu Rev Immunol 24:739–769
Paul M, Ma JK (2010) Plant-made immunogens and effective delivery strategies. Expert Rev Vaccines 9:821–833
Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, Hu D, Tappero JW, Choopanya K (2006) Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis 194:1661–1671
Rosales-Mendoza S, Alpuche-Solís AG, Soria-Guerra RE, Moreno-Fierros L, Martínez-González L, Herrera-Díaz A, Korban SS (2009) Expression of an Escherichia coli antigenic fusion protein comprising the heat labile toxin B subunit and the heat stable toxin, and its assembly as a functional oligomer in transplastomic tobacco plants. Plant J 57:45–54
Russell ND, Graham BS, Keefer MC, McElrath MJ, Self SG, Weinhold KJ, Montefiori DC, Ferrari G, Horton H, Tomaras GD, Gurunathan S, Baglyos L, Frey SE, Mulligan MJ, Harro CD, Buchbinder SP, Baden LR, Blattner WA, Koblin BA, Corey L (2004) Phase 2 study of an HIV-1 canarypox vaccine (vCP1452) alone and in combination with rgp120: negative results fail to trigger a phase 3 correlates trial. J Acquir Immune Defic Syndr 44:203–212
Saavedra R, Segura E, Leyva R, Esparza LA, López-Marín LM (2001) Mycobacterial di-O-acyl-trehalose inhibits mitogen- and antigen-induced proliferation of murine T cells in vitro. Clin Diagn Lab Immunol 8:1081–1088
Scotti N, Alagna F, Ferraiolo E, Formisano G, Sannino L, Buonaguro L, De Stradis A, Vitale A, Monti L, Grillo S, Buonaguro FM, Cardi T (2009) High-level expression of the HIV-1 Pr55gag polyprotein in transgenic tobacco chloroplasts. Planta 229:1109–1122
Scotti N, Buonaguro L, Tornesello ML, Cardi T, Buonaguro FM (2010) Plant-based anti-HIV-1 strategies: vaccine molecules and antiviral approaches. Expert Rev Vaccines 9:925–936
Soria-Guerra RE, Moreno-Fierros L, Rosales-Mendoza S (2011) Two decades of plant-based candidate vaccines: a review of the chimeric protein approaches. Plant Cell Rep 30:1367–1382
Staats HF, Nichols WG, Palker TJ (1996) Mucosal immunity to HIV-1: systemic and vaginal antibody responses after intranasal immunization with the HIV-1 C4/V3 peptide T1SP10 MN(A). J Immunol 157:462–472
Varona-Santos JT, Vazquez-Padrón RI, Moreno-Fierros L (2006) Production of a short recombinant C4V3 HIV-1 immunogen that induces strong anti-HIV responses by systemic and mucosal routes without the need of adjuvants. Viral Immunol 19:237–249
Webster DE, Thomas MC, Pickering R, Whyte A, Dry IB, Gorry PR, Wesselingh SL (2005) Is there a role for plant-made vaccines in the prevention of HIV/AIDS? Immunol Cell Biol 83:239–247
Yang ZY, Chakrabarti BK, Xu L, Welcher B, Kong WP, Leung K, Panet A, Mascola JR, Nabel GJ (2004) Selective modification of variable loops alters tropism and enhances immunogenicity of human immunodeficiency virus type 1 envelope. J Virol 78:4029–4036
Yu M, Vajdy M (2010) Mucosal HIV transmission and vaccination strategies through oral compared with vaginal and rectal routes. Expert Opin Biol Ther 10:1181–1195
Yusibov V, Streatfield SJ, Kushnir N (2011) Clinical development of plant-produced recombinant pharmaceuticals: vaccines, antibodies, and beyond. Human Vaccine 7:313–321
Zhou F, Badillo-Corona JA, Karcher D, Gonzalez-Rabade N, Piepenburg K, Borchers AM, Maloney AP, Kavanagh TA, Gray JC, Bock R (2008) High-level expression of human immunodeficiency virus antigens from the tobacco and tomato plastid genomes. Plant Biotechnol J 6:897–913
Acknowledgments
This research was funded by grants from CONACYT (102109/56980), and partially funded by a UASLP grants C08-FAI-10-6.42 to SRM and PROMEP-2010 to Bioprocess CA. Thanks are extended to Dr. Hans Ulrich Koop and Areli Herrera-Díaz for their valuable advice on plastid transformation technologies and also to Rosalba Castillo-Collazo, Benita Ortega-Berlanga, and Luzmila Martínez-González for their technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Rubio-Infante, N., Govea-Alonso, D.O., Alpuche-Solís, Á.G. et al. A chloroplast-derived C4V3 polypeptide from the human immunodeficiency virus (HIV) is orally immunogenic in mice. Plant Mol Biol 78, 337–349 (2012). https://doi.org/10.1007/s11103-011-9870-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-011-9870-1