
Abstract
Over the past decade we have witnessed the increasing sophistication of machine learning algorithms applied in daily use from internet searches, voice recognition, social network software to machine vision software in cameras, phones, robots and self-driving cars. Pharmaceutical research has also seen its fair share of machine learning developments. For example, applying such methods to mine the growing datasets that are created in drug discovery not only enables us to learn from the past but to predict a molecule’s properties and behavior in future. The latest machine learning algorithm garnering significant attention is deep learning, which is an artificial neural network with multiple hidden layers. Publications over the last 3 years suggest that this algorithm may have advantages over previous machine learning methods and offer a slight but discernable edge in predictive performance. The time has come for a balanced review of this technique but also to apply machine learning methods such as deep learning across a wider array of endpoints relevant to pharmaceutical research for which the datasets are growing such as physicochemical property prediction, formulation prediction, absorption, distribution, metabolism, excretion and toxicity (ADME/Tox), target prediction and skin permeation, etc. We also show that there are many potential applications of deep learning beyond cheminformatics. It will be important to perform prospective testing (which has been carried out rarely to date) in order to convince skeptics that there will be benefits from investing in this technique.


Similar content being viewed by others
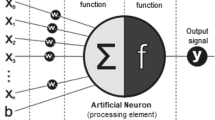
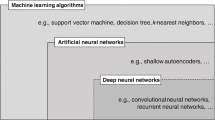

Abbreviations
- ADME/Tox:
-
Absorption, distribution, metabolism, excretion/toxicology
- AUC:
-
Area under the curve
- DILI:
-
Drug induced liver injury
- hERG:
-
Human ether a-go-go related gene
- PLGA:
-
Poly-lactide-co-glycolide
- QSAR:
-
Quantitative structure activity relationships
- SVM:
-
Support vector machines
References
Ekins S, Gupta RR, Gifford E, Bunin BA, Waller CL. Chemical space: missing pieces in cheminformatics. Pharm Res. 2010;27(10):2035–9.
Rost B, Radivojac P, Bromberg Y. Protein function in precision medicine: deep understanding with machine learning. FEBS Lett. 2016;590(15):2327–41.
Mitchell JB. Machine learning methods in chemoinformatics. Wiley Interdiscip Rev Comput Mol Sci. 2014;4(5):468–81.
Zhu H, Zhang J, Kim MT, Boison A, Sedykh A, Moran K. Big data in chemical toxicity research: the use of high-throughput screening assays to identify potential toxicants. Chem Res Toxicol. 2014;27(10):1643–51.
Clark AM, Ekins S. Open source bayesian models: 2. Mining a “big dataset” to create and validate models with ChEMBL. J Chem Inf Model. 2015;55:1246–60.
Ekins S, Clark AM, Swamidass SJ, Litterman N, Williams AJ. Bigger data, collaborative tools and the future of predictive drug discovery. J Comput Aided Mol Des. 2014;28(10):997–1008.
Ekins S, Freundlich JS, Reynolds RC. Are bigger data sets better for machine learning? Fusing single-point and dual-event dose response data for mycobacterium tuberculosis. J Chem Inf Model. 2014;54:2157–65.
Ekins S, Ecker GF, Chiba P, Swaan PW. Future directions for drug transporter modelling. Xenobiotica. 2007;37(10):1152–70.
Clark AM, Sarker M, Ekins S. New target predictions and visualization tools incorporating open source molecular fingerprints for TB Mobile 2.0. J Cheminform. 2014;6:38.
Ekins S, Clark AM, Wright SH. Making transporter models for drug-drug interaction prediction mobile. Drug Metab Dispos. 2015;43:1642–5.
Baskin II, Winkler D, Tetko IV. A renaissance of neural networks in drug discovery. Expert Opin Drug Discovery. 2016;11:785–95.
LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–44.
Burden F, Winkler D. Bayesian regularization of neural networks. Methods Mol Biol. 2008;458:25–44.
Gawehn E, Hiss JA, Schneider G. Deep learning in drug discovery. Mol Inf. 2016;35(1):3–14.
Mamoshina P, Vieira A, Putin E, Zhavoronkov A. Applications of deep learning in biomedicine. Mol Pharm. 2016;13(5):1445–54.
Chow J-F. Things to try after useR! – Part 1: Deep Learning with H2O. 2016 Aug 8th Available from: http://www.r-bloggers.com/things-to-try-after-user-part-1-deep-learning-with-h2o/.
Anon. TensorFlow. 2016 Aug 8th. Available from: https://www.tensorflow.org/.
Anon. Deeplearning4j 2016 Aug 8th. Available from: http://deeplearning4j.org/.
Novet J. Facebook open-sources its cutting-edge deep learning tools. 2016 Aug 8th. Available from: http://venturebeat.com/2015/01/16/facebook-opens-up-about-more-of-its-cutting-edge-deep-learning-tools/.
Chintala S. FAIR open sources deep-learning modules for Torch. 2016 Aug 8th. Available from: https://research.facebook.com/blog/fair-open-sources-deep-learning-modules-for-torch/.
Linn A. Microsoft releases CNTK, its open source deep learning toolkit, on GitHub. 2016 Aug 8th. Available from: http://blogs.microsoft.com/next/2016/01/25/microsoft-releases-cntk-its-open-source-deep-learning-toolkit-on-github/#sm.00013j280xp1sdctrgg21w81es5ov.
Angermueller C, Parnamaa T, Parts L, Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878.
Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2016. doi:10.1093/bib/bbw068.
Deng X, Gumm J, Karki S, Eickholt J, Cheng J. An overview of practical applications of protein disorder prediction and drive for faster, more accurate predictions. Int J Mol Sci. 2015;16(7):15384–404.
Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. 2013;53(7):1563–75.
Xu Y, Dai Z, Chen F, Gao S, Pei J, Lai L. Deep learning for drug-induced liver injury. J Chem Inf Model. 2015;55(10):2085–93.
Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure-activity relationships. J Chem Inf Model. 2015;55(2):263–74.
Putin E, Mamoshina P, Aliper A, Korzinkin M, Moskalev A, Kolosov A, et al. Deep biomarkers of human aging: application of deep neural networks to biomarker development. Aging (Albany NY). 2016;8(5):1021–33.
Di Lena P, Nagata K, Baldi P. Deep architectures for protein contact map prediction. Bioinformatics. 2012;28(19):2449–57.
Fakoor R, Ladhak F, Nazi A, Huber M. Using deep learning to enhance cancer diagnosis and classification. In: Proceeding of the 30th International conference on machine learning. Atlanta, GA: JMLR: W&CP; 2013.
Zeng T, Li R, Mukkamala R, Ye J, Ji S. Deep convolutional neural networks for annotating gene expression patterns in the mouse brain. BMC Bioinf. 2015;16:147.
Zhang S, Zhou J, Hu H, Gong H, Chen L, Cheng C, et al. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 2016;44(4):e32.
Kandaswamy C, Silva LM, Alexandre LA, Santos JM. High-content analysis of breast cancer using single-cell deep transfer learning. J Biomol Screen. 2016;21(3):252–9.
Hughes TB, Miller GP, Swamidass SJ. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent Sci. 2015;1(4):168–80.
Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: toxicity prediction using deep learning. Front Environ Sci. 2016;3:80.
Abdelaziz A, Spahn-Langguth H, Schramm K-W, Tetko IV. Consensus modeling for HTS assays using in silico descriptors calculates the best balanced accuracy in Tox21 challenge. Front Environ Sci. 2016;4:2.
Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm. 2016;13(7):2524–30.
Chen CL, Mahjoubfar A, Tai LC, Blaby IK, Huang A, Niazi KR, et al. Deep learning in label-free cell classification. Sci Rep. 2016;6:21471.
Kraus OZ, Ba JL, Frey BJ. Classifying and segmenting microscopy images with deep multiple instance learning. Bioinformatics. 2016;32(12):i52–9.
Park S, Lee SJ, Weiss E, Motai Y. Intra- and inter-fractional variation prediction of lung tumors using fuzzy deep learning. IEEE J Transl Eng Health Med. 2016;4:4300112.
Wang C, Liu J, Luo F, Tan Y. Pairwise input neural network for target-ligand interaction prediction. IEEE Int Conf Bioinf and Biomed. 2014:67-70. doi:10.1109/BIBM.2014.6999129.
Akbal-Delibas B, Farhoodi R, Pomplun M, Haspel N. Accurate refinement of docked protein complexes using evolutionary information and deep learning. J Bioinform Comput Biol. 2016;14(3):1642002.
Zawbaa HM, Szlek J, Grosan C, Jachowicz R, Mendyk A. Computational intelligence modeling of the macromolecules release from PLGA microspheres-focus on feature selection. PLoS One. 2016;11(6):e0157610.
Ekins S, Williams AJ, Xu JJ. A predictive ligand-based Bayesian model for human drug induced liver injury. Drug Metab Dispos. 2010;38:2302–8.
Clark AM, Dole K, Coulon-Spector A, McNutt A, Grass G, Freundlich JS, et al. Open source bayesian models: 1. Application to ADME/Tox and drug discovery datasets. J Chem Inf Model. 2015;55:1231–45.
Cheng T, Li Q, Wang Y, Bryant SH. Binary classification of aqueous solubility using support vector machines with reduction and recombination feature selection. J Chem Inf Model. 2011;51(2):229–36.
Low Y, Uehara T, Minowa Y, Yamada H, Ohno Y, Urushidani T, et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem Res Toxicol. 2011;24(8):1251–62.
Ekins S. Progress in computational toxicology. J Pharmacol Toxicol Methods. 2014;69(2):115–40.
Wang S, Li Y, Wang J, Chen L, Zhang L, Yu H, et al. ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol Pharm. 2012;9(4):996–1010.
Kortagere S, Chekmarev DS, Welsh WJ, Ekins S. New predictive models for blood brain barrier permeability of drug-like molecules. Pharm Res. 2008;25:1836–45.
Leong MK. A novel approach using pharmacophore ensemble/support vector machine (PhE/SVM) for prediction of hERG liability. Chem Res Toxicol. 2007;20(2):217–26.
Perryman AL, Stratton TP, Ekins S, Freundlich JS. Predicting mouse liver microsomal stability with “pruned” machine learning models and public data. Pharm Res. 2015;33:433–49.
Hou T, Wang J, Li Y. ADME evaluation in drug discovery. 8. The prediction of human intestinal absorption by a support vector machine. J Chem Inf Model. 2007;47(6):2408–15.
Clark AM, Dole K, Ekins S. Open source Bayesian models: 3. Composite models for prediction of binned responses. J Chem Inf Model. 2016;56:275–85.
Kim S, Jin D, Lee H. Predicting drug-target interactions using drug-drug interactions. PLoS One. 2013;8(11):e80129.
Unterthiner T, Mayr A, Klambauer G, Hochreiter S. Toxicity prediction using deep learning. Available from: https://arxiv.org/pdf/1503.01445.pdf.
Baba H, Takahara J, Mamitsuka H. In silico predictions of human skin permeability using nonlinear quantitative structure-property relationship models. Pharm Res. 2015;32(7):2360–71.
Baba H, Takahara J, Yamashita F, Hashida M. Modeling and prediction of solvent effect on human skin permeability using support vector regression and random forest. Pharm Res. 2015;32(11):3604–17.
Zheng X, Ekins S, Raufman JP, Polli JE. Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharm. 2009;6(5):1591–603.
Diao L, Ekins S, Polli JE. Quantitative structure activity relationship for inhibition of human organic cation/carnitine transporter. Mol Pharm. 2010;7:2120–30.
Dong Z, Ekins S, Polli JE. Structure-activity relationship for FDA approved drugs as inhibitors of the human sodium taurocholate cotransporting polypeptide (NTCP). Mol Pharm. 2013;10(3):1008–19.
You H, Lee K, Lee S, Hwang SB, Kim KY, Cho KH, et al. Computational classification models for predicting the interaction of compounds with hepatic organic ion importers. Drug Metab Pharmacokinet. 2015;30(5):347–51.
de Cerqueira Lima P, Golbraikh A, Oloff S, Xiao Y, Tropsha A. Combinatorial QSAR modeling of P-glycoprotein substrates. J Chem Inf Model. 2006;46(3):1245–54.
Xue Y, Yap CW, Sun LZ, Cao ZW, Wang JF, Chen YZ. Prediction of P-glycoprotein substrates by a support vector machine approach. J Chem Inf Comput Sci. 2004;44(4):1497–505.
Xu C, Cheng F, Chen L, Du Z, Li W, Liu G, et al. In silico prediction of chemical Ames mutagenicity. J Chem Inf Model. 2012;52(11):2840–7.
Hansen K, Mika S, Schroeter T, Sutter A, ter Laak A, Steger-Hartmann T, et al. Benchmark data set for in silico prediction of Ames mutagenicity. J Chem Inf Model. 2009;49(9):2077–81.
Moss GP, Shah AJ, Adams RG, Davey N, Wilkinson SC, Pugh WJ, et al. The application of discriminant analysis and Machine Learning methods as tools to identify and classify compounds with potential as transdermal enhancers. Eur J Pharm Sci. 2012;45(1-2):116–27.
Vock DM, Wolfson J, Bandyopadhyay S, Adomavicius G, Johnson PE, Vazquez-Benitez G, et al. Adapting machine learning techniques to censored time-to-event health record data: A general-purpose approach using inverse probability of censoring weighting. J Biomed Inform. 2016;61:119–31.
Chia CC, Rubinfeld I, Scirica BM, McMillan S, Gurm HS, Syed Z. Looking beyond historical patient outcomes to improve clinical models. Sci Transl Med. 2012;4(131):131ra149.
Rochefort CM, Verma AD, Eguale T, Lee TC, Buckeridge DL. A novel method of adverse event detection can accurately identify venous thromboembolisms (VTEs) from narrative electronic health record data. J Am Med Inform Assoc. 2015;22(1):155–65.
Degardin K, Guillemain A, Guerreiro NV, Roggo Y. Near infrared spectroscopy for counterfeit detection using a large database of pharmaceutical tablets. J Pharm Biomed Anal. 2016;128:89–97.
Khamis MA, Gomaa W, Ahmed WF. Machine learning in computational docking. Artif Intell Med. 2015;63(3):135–52.
Sushko I, Novotarskyi S, Korner R, Pandey AK, Rupp M, Teetz W, et al. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des. 2011;25(6):533–54.
Walker T, Grulke CM, Pozefsky D, Tropsha A. Chembench: a cheminformatics workbench. Bioinformatics. 2010;26(23):3000–1.
Ekins S, Hohman M, Bunin BA. Pioneering use of the cloud for development of the collaborative drug discovery (cdd) database. In: Ekins S, Hupcey MAZ, Williams AJ, editors. Collaborative computational technologies for biomedical research. Hoboken: Wiley; 2011.
Ekins S, Litterman NK, Lipinski CA, Bunin BA. Thermodynamic proxies to compensate for biases in drug discovery methods. Pharm Res. 2016;33(1):194–205.
Lienard P, Gavartin J, Boccardi G, Meunier M. Predicting drug substances autoxidation. Pharm Res. 2015;32(1):300–10.
Fagerberg JH, Karlsson E, Ulander J, Hanisch G, Bergstrom CA. Computational prediction of drug solubility in fasted simulated and aspirated human intestinal fluid. Pharm Res. 2015;32(2):578–89.
Kingsley LJ, Wilson GL, Essex ME, Lill MA. Combining structure- and ligand-based approaches to improve site of metabolism prediction in CYP2C9 substrates. Pharm Res. 2015;32(3):986–1001.
Wang W, Kim MT, Sedykh A, Zhu H. Developing enhanced blood-brain barrier permeability models: integrating external bio-assay data in QSAR modeling. Pharm Res. 2015;32(9):3055–65.
Hatanaka T, Yoshida S, Kadhum WR, Todo H, Sugibayashi K. In silico estimation of skin concentration following the dermal exposure to chemicals. Pharm Res. 2015;32(12):3965–74.
Anon. Special report: the return of the machinery question. In: The Economist; 2016 June 25th. Available from: http://www.economist.com/news/special-report/21700761-after-many-false-starts-artificialintelligence-has-taken-will-it-cause-mass.
Ekins S, Reynolds R, Kim H, Koo M-S, Ekonomidis M, Talaue M, et al. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem Biol. 2013;20:370–8.
Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, et al. Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening. J Chem Inf Model. 2013;53(2):475–92.
Whelan KE, King RD. Intelligent software for laboratory automation. Trends Biotechnol. 2004;22(9):440–5.
Tetko IV, Bruneau P, Mewes HW, Rohrer DC, Poda GI. Can we estimate the accuracy of ADME-Tox predictions? Drug Discov Today. 2006;11(15-16):700–7.
Fourches D, Muratov E, Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model. 2010;50(7):1189–204.
Williams AJ, Ekins S, Tkachenko V. Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov Today. 2012;17:685–701.
Vracko M, Bandelj V, Barbieri P, Benfenati E, Chaudhry Q, Cronin M, et al. Validation of counter propagation neural network models for predictive toxicology according to the OECD principles: a case study. SAR QSAR Environ Res. 2006;17(3):265–84.
Ekins S, Wood J. Incentives for Starting Small Companies Focused on Rare and Neglected Diseases. Pharm Res. 2016;33:809–15.
Ponder EL, Freundlich JS, Sarker M, Ekins S. Computational models for neglected diseases: gaps and opportunities. Pharm Res. 2014;31(2):271–7.
Murnane K. What is deep learning and how is it useful? Forbes. Available from: http://www.forbes.com/sites/kevinmurnane/2016/04/01/what-is-deep-learning-and-how-is-it-useful/#715d1eaf10f0.
Murnane K. Thirteen companies that use deep learning to produce actionable results. Forbes. Available from: http://www.forbes.com/sites/kevinmurnane/2016/04/01/thirteen-companies-that-use-deep-learning-to-produce-actionable-results/#4e710eb07967.
Tetko IV, M Lowe D, Williams AJ. The development of models to predict melting and pyrolysis point data associated with several hundred thousand compounds mined from PATENTS. J Cheminform. 2016;8:2.
Tetko IV, Novotarskyi S, Sushko I, Ivanov V, Petrenko AE, Dieden R, et al. Development of dimethyl sulfoxide solubility models using 163,000 molecules: using a domain applicability metric to select more reliable predictions. J Chem Inf Model. 2013;53(8):1990–2000.
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44(D1):D1202–13.
ACKNOWLEDGMENTS AND DISCLOSURES
Dr. Alex M. Clark and Dr. Peter W. Swaan are kindly acknowledged for useful discussions on this topic. SE is founder and owner of Collaborations Pharmaceuticals, Inc. he was a consultant for Collaborative Drug Discovery, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was partially supported by Award Number 9R44TR000942-02 “Biocomputation across distributed private datasets to enhance drug discovery” from the NIH National Center for Advancing Translational Sciences.
Rights and permissions
About this article

Cite this article
Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm Res 33, 2594–2603 (2016). https://doi.org/10.1007/s11095-016-2029-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-016-2029-7