
ABSTRACT
Purpose
To investigate the in vitro release of octreotide acetate, a somatostatin agonist, from microspheres based on a hydrophilic polyester, poly(D,L-lactide-co-hydroxymethyl glycolide) (PLHMGA).
Methods
Spherical and non-porous octreotide-loaded PLHMGA microspheres (12 to 16 μm) and loading efficiency of 60–70% were prepared by a solvent evaporation. Octreotide release profiles were compared with commercial PLGA formulation (Sandostatin LAR®); possible peptide modification with lactic, glycolic and hydroxymethyl glycolic acid units was monitored.
Results
PLHMGA microspheres showed burst release (~20%) followed by sustained release for 20–60 days, depending on the hydrophilicity of the polymer. Percentage of released loaded peptide was high (70–90%); > 60% of released peptide was native octreotide. PLGA microspheres did not show peptide release for the first 10 days, after which it was released in a sustained manner over the next 90 days; > 75% of released peptides were acylated adducts.
Conclusions
PLHMGA microspheres are promising controlled systems for peptides with excellent control over release kinetics. Moreover, substantially less peptide modification occurred in PLHMGA than in PLGA microspheres.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Microspheres of biodegradable polymers with poly(D,L-lactide-co-glycolide) (PLGA) as main representative have been widely studied for the prolonged release of bioactive peptides (1–7). However, one of the issues in peptide delivery using PLGA formulations is the formation of peptide adducts as a result of acylation with lactic and glycolic units (8,9). Acylation of, particularly, lysine residues in peptides is catalyzed by the low pH that is generated inside degrading PLGA microspheres due to the accumulation of degradation products, i.e. lactic and glycolic acid and their oligomers (10–14). The investigated options to avoid chemical derivatization of peptides entrapped in PLGA matrices, such as PEGylation and co-encapsulation of water-soluble divalent cationic salts, resulted in less acylation (15–17). However, it is obvious that these approaches can not be generally applied, and consequently other options need further exploration. Besides acylation, incomplete and difficult-to-tailor release of peptides from PLGA microparticles are other challenges hampering widespread clinical application of these peptide formulations (18,19). Within our department, a new hydrophilic polyester, poly(lactide-co-hydroxymethyl glycolide) (PLHMGA), has been developed (20,21). Protein-loaded PLHMGA microspheres based on copolymers with different ratios of D,L-lactide and hydroxymethyl glycolide (HMG) (75/25, 65/35 and 50/50) were prepared using a double-emulsion solvent evaporation method. Degradation of the microspheres and release of model proteins (lysozyme and BSA) were investigated. It was demonstrated that the release of model proteins was governed by degradation of the microspheres and that duration of the release could be tailored from 2 weeks to 2 months (22,23). Spectroscopic and chromatographic analysis as well as bioactivity measurements (lysozyme) showed that the released proteins retained their structural integrity.
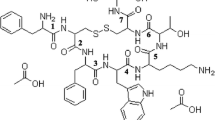
In the present study we investigated the suitability of PLHMGA microspheres for the release of a therapeutic peptide, octreotide (structure shown in Fig. 1). This synthetic peptide mimics the peptide hormone somatostatin and has received FDA approval for the treatment of acromegaly (a condition in which excess amount of growth hormone is produced from the anterior pituitary) and gastroenteropancreatic neuroendocrine tumors (24,25). Because of its poor pharmacokinetics (plasma half-life of about 100 min following iv and sc injections (26,27)), sustained release octreotide formulations based on PLGA microspheres have been developed. One formulation named Sandostatin LAR® made with a PLGA-glucose star polymer is commercially available and used in clinics for the treatment of acromegaly and gastrointestinal tumors (28,29). However, previous studies with octreotide-PLGA microspheres showed an incomplete and difficult-to-tailor release (30). Importantly, HPLC and mass spectrometric analysis demonstrated the presence of octreotide derivatives (lactoyl and glycoyl adducts) in the release samples (17,30). It was hypothesized that a nucleophilic attack of the primary amine groups present in the N-terminus and lysine residue of the peptide on the electrophilic carbonyl ester groups present in the PLGA backbone resulted in peptide acylation (9,31).

Structure of octreotide acetate; acylation most likely occurs at positions 1 and 2 (http://www.chemblink.com/products/83150-76-9.htm).
Due to steric factors, the nucleophilic attack of octreotide more readily occurred on glycolic acid rather than lactic acid units (9). Because PLHMGA lacks glycolic acid units, we hypothesized that octreotide encapsulated in PLHMGA microspheres is less susceptible to acylation. Moreover, the extra hydroxyl groups in PLHMGA increases the water-absorbing capacity of the degrading polymer matrix and facilitates the release of acid degradation products, thereby preventing a pH drop (manuscript in preparation). It is further anticipated that, as previously shown for BSA (23), the release of the peptide can be tailored by the degradation kinetics of the polymer, which in turn depends on the copolymer composition. Therefore, the aim of this study was to investigate PLHMGA microspheres of different composition for the controlled release of octreotide and compare the release kinetics with octreotide release from Sandostatin LAR®. Possible modification of the released peptide was investigated by HPLC and MALDI-TOF analysis.
MATERIALS AND METHODS
Materials
O-Benzyl-L-serine was purchased from Senn Chemicals AG (Dielsdorf, Switzerland). D,L-lactide was obtained from Purac, the Netherlands. N,N´-Dimethylformamide and methyl-tert-butyl ether (MTBE), peptide grade dichloromethane (DCM), methanol, chloroform and tetrahydrofurane (THF) were purchased from Biosolve (Valkenswaard, the Netherlands). Benzyl alcohol was obtained from Merck (Darmstadt, Germany). Toluene (Acros, Geel, Belgium) was distilled from P2O5 and stored over 3 Å molecular sieves under argon. N,N´-Dimethylaminopyridine (DMAP) and sodium azide (NaN3, 99%) were purchased from Fluka (Zwijndrecht, the Netherlands). Disodium hydrogen phosphate dihydrate (Na2HPO .4 2H2O) and sodium dihydrogen phosphate monohydrate (NaH2PO .4 H2O) were obtained from Merck (Darmstadt, Germany). Polyvinyl alcohol (PVA; Mw 30,000-70,000; 88% hydrolyzed) and tin (II) 2-ethylhexanoate (SnOct2) were from Sigma-Aldrich, Inc., USA. Pd/C (Palladium, 10 wt. % on activated carbon, Degussa type E101 NE/W) was purchased from Aldrich, Zwijndrecht, the Netherlands. Octreotide acetate (H2N-D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-ol; MW = 1,019 Da) was obtained from Feldan-bio (Quebec, Canada). Sandostatin LAR® (Batch No. S0090) was obtained from Novartis, Basel, Switzerland. According to the supplier, the loading capacity of this formulation was 20 mg peptide per 500 mg microspheres. Unless otherwise stated, all chemicals were used as received.
Synthesis of Copolymers of 3S-(benzyloxymethyl)-6S-methyl-1,4-dioxane-2,5-dione with D,L-lactide
3S-(benzyloxymethyl)-6S-methyl-1,4-dioxane-2,5-dione (BMMG) was synthesized as described before by Leemhuis et al. (20). Copolymers of D,L-lactide and BMMG with different monomer ratios (75/25, 65/35 and 50/50% D,L-lactide/BMMG mol/mol) were synthesized by melt copolymerization (Fig. 2) (20).

Synthesis of hydrophilic aliphatic polyesters based on lactic acid and glycolic acid with pendant hydroxyl groups, poly(lactic-co-hydroxymethyl glycolic acid) (20).
As a representative example, for the 75/25 copolymer, D,L-lactide (1,253 mg, 8.66 mmol) and BMMG (726 mg, 2.87 mmol) were transferred into a dried Schlenk tube under a dry nitrogen atmosphere. Benzyl alcohol (12.54 mg; 37 μl of a 352 mg/ml toluene stock solution) and SnOct2 (23.5 mg; 71 μl of a 330 mg/ml toluene stock solution) were added as initiator and catalyst, respectively. Toluene was removed under vacuum, and the tube was sealed and immersed into a 130°C oil bath for 16 h while stirring. After cooling to room temperature, the obtained copolymer was dissolved in chloroform and subsequently precipitated in cold methanol. The precipitate was collected by filtration and vacuum dried to yield 1.86 g of poly(D,L-lactic acid-ran-benzyloxymethyl glycolic acid) (PLBMGA) as a white solid. The copolymer was dissolved in 300 ml distilled THF, and 10% w/w Pd/C was added. The solution was stirred under a hydrogen atmosphere for 16 h to remove the benzyl protecting group, and the catalyst was subsequently removed by Hyflo filter. Evaporation of THF yielded 1.5 g of poly(D,L-lactide-co-hydroxymethyl glycolide) (PLHMGA). Copolymers of 65/35 and 50/50 (mol/mol D,L-lactide and BMMG) were synthesized using the same method by adjusting the amount of BMMG and D,L-lactide in the feed.
Polymer Characterization
1H NMR measurements of PLBMGA and PLHMGA dissolved in CDCl3 were performed at 298 K on a Varian Gemini-300 MHz spectrometer. Molecular weights of the synthesized polymers were determined by GPC analysis (Waters Alliance equipped with a Waters 2695 separating module and a Waters 2414 refractive index detector). Two PL-gel 5 μm Mixed-D columns fitted with a guard column .1pt?>(Polymer Labs, M w range 0.2-400 kDa) were used. Calibration was performed with polystyrene standards using THF as the mobile phase (1 ml/min). PLBMGA/PLHMGA was dissolved in THF (5% w/v), and 50 μl was injected into the column (22). The thermal properties of the protected and deprotected copolymers were measured using differential scanning calorimetry (TA instrument, Q2000) as described before (23). The second cycle was used to determine the glass transition temperature (Tg) of synthesized polymers.
Preparation of Octreotide Microspheres
Microspheres were prepared using a solvent evaporation technique (22,32). In an optimized protocol, 50 μl of an octreotide solution in reversed osmosis (RO) water (200 mg/ml) was emulsified with 500 μl of dichloromethane solution of PLHMGA (220 mg, 25% w/w) by using an IKA homogenizer (IKA Labortechnik Staufen, Germany) for 30 s at the highest speed (30,000 rpm) to get the primary emulsion. Next, 500 μl of a PVA solution (1% w/w in 30 mM phosphate buffer, pH 7.4) was added slowly, and the mixture was vortexed for 30 s at 30,000 rpm. The obtained w/o/w emulsion was then transferred into a 5 ml PVA solution 0.5% (w/w) in 30 mM phosphate buffer pH 7.4 while stirring. Continuous stirring at room temperature for 2 h resulted in extraction/evaporation of DCM. Finally, hardened microspheres were collected by centrifugation (Laboratory centrifuge, 4 K 15 Germany) at 3,000 g for 3 min, subsequently washed 3 times with 100 ml RO water and freeze dried overnight. The dried microspheres were stored at −25°C. Microspheres were also prepared with other formulation and processing parameters (see Supplementary Material).
Microspheres Characterization
The microspheres’ size distribution was analyzed by an Accusizerth 780 (Optical particle sizer, Santa Barbara, California, USA). Results are reported as volumetric mean diameter.
The morphology of the microspheres after freeze-drying was analyzed by scanning electron microscopy using a PhenomTM SEM (FEI Company, the Netherlands). The samples were mounted onto a 12 mm diameter aluminum specimen stub (Agar Scientific Ltd., England) using double-sided adhesive tape and were sputter-coated with platinum.
The octreotide loading of the microspheres (PLHMGA as well as the PLGA/Sandostatin LAR® formulation) was determined using an HPLC method according to Wang et al. (33). In detail, about 10 mg of PLHMGA microspheres were dissolved in 0.5 ml THF with gentle shaking. Thereafter, 2 ml of diluent (0.2% w/v glacial acetic acid, 0.2% w/v sodium acetate and 0.7% w/v sodium chloride in water) was added to precipitate the polymer. Next, the mixture was incubated at room temperature for 20 min, and the precipitated polymer was spun down by centrifugation at 5,000 g for 1 min. The protein content in the supernatant was measured by high performance liquid chromatography (HPLC) using a Prosphere HP C4-300 column. A gradient elution method was used with a mobile phase A (95% H2O, 5% ACN and 0.1% TFA) and a mobile phase B (100% ACN and 0.1% TFA). The eluent linearly changed from 100% to 40% A in 20 min with a flow rate of 1 ml/min. Octreotide standards (5–200 μg/ml, 20 μl injection volume) were used for calibration, and detection was done at 210 nm. Loading efficiency (LE) of octreotide in microspheres is reported as the encapsulated peptide divided by the total amount of peptide used for encapsulation. Loading capacity (LC) is expressed as the encapsulated amount of octreotide divided by the total dry weight of the microspheres.
In Vitro Release Studies
The release of octreotide from PLHMGA and Sandostatin LAR® microspheres was studied in PBS (0.033 M NaH2PO4, 0.066 M Na2HPO4, 0.056 M NaCl and 0.05% (w/w) NaN3, pH 7.4). About 30 mg of microspheres was suspended into 1.5 ml of PBS buffer in eppendorf tubes and incubated at 37°C under mild agitation using a circular mixer (ASSISTANT RM 5). At the different time points, the dispersion was centrifuged (3,000 g, 3 min), and 1 ml of the supernatant was replaced with 1 ml of fresh buffer. The microspheres were resuspended by gentle shaking, and the dispersion was incubated at 37°C. The peptide concentration in the release samples was measured by HPLC as described in section 2.5.3.
MALDI-TOF Analysis
Different release samples were analyzed using MALDI-TOF (Applied Biosystems 4700, Foster City, CA, equipped with a 200 Hz Nd:YAG laser operating at 355 nm). Samples were diluted to 5 μg/ml in a solution of 3 mg/ml α-cyano-4-hydroxycinnamic acid dissolved in 1:1 acetonitrile/MilliQ (v/v) containing 0.1% (v/v) trifluoro acetic acid. The sample (about 1 μl) was spotted onto a stainless steel matrix-assisted laser desorption/ionization (MALDI) plate. Analysis was performed in a reflectron positive ion mode, and, typically, 3,000 shots/spectrum were acquired in the MS mode.
RESULTS AND DISCUSSION
Synthesis and Characterization of PLHMGA with Different Copolymer Composition
Benzyl protected hydroxymethyl glycolide (BMMG) and D,L-lactide were copolymerized by ring opening polymerization at different feed ratios (75/25, 65/35 and 50/50 mol D,L-lactide/mol BMMG) in the melt at 130°C using BnOH and SnOct2 as initiator and catalyst, respectively. The protected copolymers (PLBMGA) were obtained in high yields (>90%), and NMR analysis showed that the copolymer composition closely matched that of the feed (Table I). DSC analysis showed that the copolymers were fully amorphous with a Tg ranging from 33 to 41°C (Table I), which is in agreement with previous findings (20,22,23). NMR analysis showed that catalytic hydrogenation of PLBMGA resulted in quantitative removal of the protecting benzyl groups to obtain PLHMGAs in a high yield. As for the protected polymers, PLHMGA of different composition were fully amorphous with a Tg of 47–49°C (Table I). GPC analysis showed that the number average molecular weights of synthesized polymers before and after deprotection ranged from 13–24 kg/mol. The molecular weight distributions were similar (Mw/Mn ~2) to those of polyesters synthesized by ring opening polymerization of lactide/glycolide in the melt (34).
Preparation and Characterization of Octreotide-Loaded Microspheres
Octreotide-loaded microspheres were prepared using a double emulsion/solvent evaporation technique as described in section 2.4. The characteristics of the microspheres are summarized in Table II.
Table II shows that the PLHMGA microspheres had about the same size that ranged from 12–16 μm. The effect of several formulation parameters, which are known to have a possible influence on the loading efficiency, was investigated (polymer concentration (35), volume of the protein solution (36), addition of salt to the continuous phase (37), volume of continuous phase (38), use of methanol as solvent for octreotide (o/w method) (8) and buffering the pH of the PVA phase at pH 7.4). The results presented in the Supplementary Material show that the LE was low (<30%) except for the formulation that was prepared with a neutralized external phase (LE was 50–60%, Table II). Likely, the driving force for extraction of octreotide from the internal to the external aqueous phase during formation of the particles is low at pH 7.4 (which is close to the pI of octreotide (8.5)) because the aqueous solubility of the peptide is low, resulting in an enhanced loading efficiency and capacity. The LE was independent of copolymer composition and is in agreement with previous findings showing around the same LE of octreotide in PLGA microspheres (33). The LC of the Sandostatin LAR® was 4.1 ± 0.1, which is within the experimental error equal to the load as specified by the supplier (20 mg peptide per 500 mg formulation). SEM analysis of the different microspheres of Table II showed that the particles are spherical and essentially non-porous (Fig. 3) and the average size by SEM corresponded well with that of accusizer measurements (Table II). SEM analysis of Sandostatin LAR® shows particles with a bigger size than the PLHMGA microspheres, in agreement with accusizer measurements (average size around 65 μm, Table II). Besides, SEM analysis also showed the presence of some smaller irregularly shaped particles in Sandostatin LAR® (Fig. 3d).

SEM analysis of microspheres: (a) PLHMGA 75/25, (b) PLHMGA 65/35, (c) PLHMGA 50/50 and (d) Sandostatin LAR® (note the difference in scale bar between (d) and (a–c).
In Vitro Release of Octreotide-Loaded PLGA/PLHMGA Microspheres
The HPLC chromatogram of octreotide in PBS (pH 7.4) incubated at 37°C for 21 days showed a sharp peak with a retention time around 10 min (Fig. 4a). No other peaks were detected, demonstrating that octreotide has a good stability under the conditions applied for evaluation of the release of the different octreotide formulations. However, octreotide released from PLHMGA and PLGA microspheres showed extra peaks with longer retention times (11–12 min) originating from acylated octreotide adducts (see MALDI-TOF Analysis section) as also observed by Murty et al. (30). It is remarked that the extent of peptide modification is more pronounced after being released from PLGA than from PLHMGA microspheres (compare Fig. 4b, c). At the end of release study when the microspheres were fully degraded (day 95 for PLGA microspheres and day 60 for PLHMGA 65/35 microspheres, respectively), mainly acylated peptide was released from PLGA microspheres, whereas mainly native octreotide was released from PLHMGA microspheres (Supplementary Material 2 and 3, respectively). It should be stressed that acylated octreotide adducts are formed during degradation of the microspheres and not during their preparation, since only native peptide was detected with HPLC when the loading capacity of the different microspheres was determined (data not shown).

HPLC chromatograms of octreotide after 21 days incubation in PBS pH 7.4 at 37°C (a) and after release from microspheres of PLHMGA (65/35, b) and PLGA (c) after 21 days.
Figure 5 shows the in vitro release of octreotide from PLGA microspheres in PBS pH 7.4 during 95 days. After 95 days the PLGA microspheres were fully degraded, in accordance to what has been reported by Spenlehauer et al. (39). The formulation showed no burst release, and after 10–15 days peptide release started, which continued for the next 80 days until a plateau value was reached at 85% of the octreotide loading. The incomplete release can be due to the formation of insoluble peptide residues (not visible with naked eye) or to adsorption of the peptide to surfaces with which it has been brought into contact. HPLC analysis (see Fig. 4) makes it possible to differentiate between octreotide that is released in its native form and in acylated forms. Of the released peptide, 25% was unmodified octreotide, whereas the remaining 75% was acylated products (Fig. 5, Table III).

In vitro release of octreotide from PLGA (the results are from one study); (∆) native octreotide, (▲) acylated octreotide and (○) total octreotide (native and acylated octreotide adducts). The percentage of release is based on the LC of PLGA microspheres detected by HPLC (Table II).
The release of octreotide from microspheres based on PLHMGA of different hydrophilicity (different copolymer composition (75/25, 65/35 and 50/50 mol/mol D,L-lactide and BMMG (after deprotection: HMG)) was studied. The results are shown in Fig. 6 and in the Supplementary Material 4; the data are summarized in Table III. Figure 6 shows the release of octreotide from microspheres of PLHMGA 65/35. It appears that after a burst (around 20% of the loading), which is probably caused by nanopores present in the microspheres that are not detected with SEM, octreotide was released in a sustained manner for 60 days, and almost quantitative release (80–90%) was obtained. Although also acylated forms of the peptide were released, more than 65% of the released amount was native octreotide, while PLGA microspheres mainly released acylated octreotide, (Table III). Importantly, octreotide-loaded PLHMGA microspheres were fully degraded after 35 days, in line with our previous study (23). The explanation for the continuation of the release after visible microsphere degradation will be given below (vide infra).

In vitro release of octreotide from PLHMGA microspheres (65/35) in PBS pH 7.4 at 37°C: (▲) acylated octreotide, (∆) native octreotide and (○) total octreotide release (native and acylated octreotide adducts) (n = 3).
Microspheres of copolymer 50/50 showed, after a burst (around 20% of the loaded amount), an almost zero order release of the peptide for 20 days until 85% of the loaded amount was released (of which 70% is native octreotide and the remaining 30% is in the form of acylated peptide; Supplementary Material 4 and Table III). Complete degradation of the microspheres, occurred in 20 days. Octreotide release from microspheres of the more slowly degrading copolymer 75/25 (Supplementary Material 5) was characterized by a burst (around 20% of the loaded amount) followed by a sustained release for more than 60 days. In line with the results of PLHMGA 65/35 and 50/50 microspheres, around 70% of the released peptide was native octreotide, whereas 30% was acylated adducts. PLHMGA 75/25 microspheres degraded completely in 55 days. There is a tendency that the total released amount of peptide decreases with increasing degradation time of the particles. This might be due to the presence of polymer-peptide conjugates in the release medium that are not detected with the HPLC method (see Fig. 7 caption). Also, adsorption of the peptide to surfaces to which it has been exposed for longer times with increasing degradation time might be responsible for this observation.

In our previous studies, the release of model proteins lysozyme and BSA from microspheres of copolymer 50/50, 65/35 and 75/25 was reported. It was concluded that the release of these proteins was essentially governed by degradation of the polymer matrices (22,23). The present results show that degradation governs to some extent the release of the peptide as well (the faster the degradation of the particles, the faster its release). Probably also diffusion plays a role, given the observation that, after the burst, the PLHMGA microspheres released the peptide without delay (as opposed to the protein delivery studies). Obviously, the hydroxyl groups of the polymer give sufficient water absorption to release the peptide by diffusion.
In the present study, we observed that after complete degradation of the microspheres (at day 35 for the PLHMGA 65/35 microspheres, Fig. 6) the release of octreotide still continued (from 62% of the loading at day 35 to 80% at day 60). This can be explained as follows. The reaction of the copolymer and a nucleophile (e.g. lysine residue) of the peptide results in the formation of a peptide-polymer conjugate (step 1, Fig. 7). Likely, these polymer-peptide conjugates have either limited solubility or are not detected by the used HPLC method (30) due to their heterogeneous character. The peptide-polymer conjugates may still be present at the time the microspheres are fully degraded and are slowly converted by hydrolysis during the next days into peptide-LA/HMGA adducts (Fig. 7, step 2), which are detected with HPLC, explaining the apparent increase in release after degradation of the microspheres (the apparent increase in total percentage of release is thus due to acylated peptide adducts). The observation that after full microspheres’ degradation the amount of released native peptide indeed hardly increased supports the above-mentioned explanation.
MALDI-TOF Analysis
The structure of the released octreotide adducts was investigated by MALDI-TOF analysis. Figure 8a shows the mass spectrum of the native octreotide with only peaks corresponding with octreotide H + (1,019 m/z) and that of the Na-adduct (1,041 m/z). Figure 8b and c show the mass spectra of octreotide released after 21 days from PLGA and PLHMGA microspheres, respectively. Clearly, besides native octreotide, also peaks with higher masses were detected. In agreement with HPLC analysis (vide supra), more extensive peptide derivatization is seen in octreotide released from PLGA than from PLHMGA microspheres. MALDI-TOF spectra of the octreotide released from microspheres based on PLHMGA of different copolymer composition were similar.

Mass spectrum of octreotide acetate after incubation of 21 days in PBS at 37°C (a), after 21 days of release from Sandostatin LAR® (b) and PLHMGA microspheres of copolymer 65/35 (c).
The attribution of the MALDI-TOF MS peaks to different octreotide derivatives is summarized in Table IV. The masses of the observed peaks of octreotide released from PLGA microspheres can be fully ascribed to peptide modified with lactic (octreotide-LA) and glycolic acid (octreotide-GA) units. These products are formed as suggested by Na et al. (15) and Zhang et al. (41). The relative amounts of octreotide-GA (peak numbers 3 and 4, Fig. 8b) was substantially greater (height ratio about 7/1) than octreotide-LA (peak numbers 5 and 6, Fig. 8b). This can be explained because the nucleophilic attack is more hindered with lactic acid monomers as compared with glycolic acid monomers (Fig. 9) (8).

Proposed explanation of reduced reactivity of nucleophilic species with lactic acid monomers (8).
No octreotide-GA was detected in octreotide released from PLHMGA microspheres due to the lack of glycolic acid units in PLHMGA, while minor amounts of octreotide-HMGA were observed instead. The ratio of octreotide-LA to octreotide-HMGA was 1.6, 1.2 and 1.3 for copolymer 75/25, 65/35 and 50/50, respectively. The 50/50 copolymer contains 75% lactic acid units and 25% HMGA units (Fig. 2). This means that if both units are equally reactive with the peptide, a 3/1 ratio octreotide-LA to octreotide-HMGA can be expected (for the peptide released from the other PLHMGA microspheres, this ratio would be even higher). Further, it should be mentioned that in time the degrading polymers become richer in lactate content (21,22), which would point to an even higher expected ratio of octreotide-LA to octreotide-HMGA. It is therefore concluded that acylation of octreotide preferentially occurs by reaction with the HMGA esters. Likely, this monomer unit due to its hydroxyl group is more hydrated than the lactic acid units in the polymer chain, favoring reaction with the peptide.
Based on the results (Fig. 5, peptide release from PLGA, Fig. 6 peptide release from PLHMGA, Fig. 8 and Table IV), it can be concluded that less peptide acylation occurs during release from PLHMGA microspheres compared to that of PLGA microspheres. Three factors might contribute to the lower extent of peptide acylation in degrading PLHMGA microspheres. First, as mentioned above, the absence of the more reactive glycolic acid units in PLHMGA slows down the acylation reaction (9). Second, the increased degradation rate promotes the removal of the HMGA units and thus reduces the adduct formation. Third, it has been shown that the low pH generated inside degrading PLGA matrices catalyzes the formation of acylated peptide adducts (40). However, less acidification of PLHMGA matrices occurs during degradation compared to that of PLGA matrices (manuscript in preparation), likely due to the more hydrophilic character of degrading PLHMGA matrices which favors the extraction of formed water-soluble acidic degradation products.
CONCLUSIONS
Our data demonstrate that the release of octreotide from PLHMGA microspheres can be tailored by the copolymer composition and outperformed that of the commercial octreotide formulation Sandostatin LAR®. Importantly, substantially less acylated octreotide adducts were formed in degrading PLHMGA microspheres than in Sandostatin LAR® due to the lack of glycolic acid and less acidification. To further decrease the acylation, PEGylation of octreotide or co-encapsulation of excipients which can prevent the nucleophilic attack of a peptide to the polymer backbone (17,41) are logical options that warrant further investigation.
REFERENCES
Mehta RC, Jeyanthi R, Calls S, Thanoo BC, Burton KW, DeLuca PP. Biodegradable microspheres as depot system for patenteral delivery of peptide drugs. J Control Release. 1994;29:375–84.
Calis S, Jeyanthi R, Tsai T, Mehta RC, DeLuca PP. Adsorption of salmon calcitonin to PLGA microspheres. Pharm Res. 1995;12:1072–6.
Yadav SC, Kumari A, Yadav R. Development of peptide and protein nanotherapeutics by nanoencapsulation and nanobioconjugation. Peptides. 2011;32:173–87.
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(d, l-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125:193–209.
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170–83.
Du L, Mei X, Wang C, Li X, Zhang F, Jin Y. In-vitro/in-vivo studies of the biodegradable poly-(d, l-lactide-co-glycolide) microspheres of a novel luteinizing hormone-releasing hormone antagonist for prostate cancer treatment. Anti-Cancer Drugs. 2011;22:262–72.
Dong WY, Körber M, López EV, Bodmeier R. Stability of poly(d, l-lactide-co-glycolide) and leuprolide acetate in in-situ forming drug delivery systems. J Control Release. 2006;115:158–67.
Murty SB, Thanoo BC, Wei Q, DeLuca PP. Impurity formation studies with peptide-loaded polymeric microspheres: Part I. In vivo evaluation. Int J Pharm. 2005;297:50–61.
Murty S, Goodman J, Thanoo B, DeLuca P. Identification of chemically modified peptide from poly(D, L-lactide-co-glycolide) microspheres under in vitro release conditions. AAPS Pharm Sci Tech. 2003;4:392–405.
Kang J, Schwendeman SP. Comparison of the effects of Mg(OH)2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(D, L-lactide-co-glycolide) implants. Biomaterials. 2002;23:239–45.
Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17:100–6.
Tia E, Jichao K, Steven PS, John FC. BSA degradation under acidic conditions: a model for protein instability during release from PLGA delivery systems. J Pharm Sci-us. 2006;95:1626–39.
Shenderova A, Ding AG, Schwendeman SP. Potentiometric method for determination of microclimate pH in poly(lactic-co-glycolic acid) films. Macromolecules. 2004;37:10052–8.
Ding AG, Shenderova A, Schwendeman SP. Prediction of microclimate pH in poly(lactic-co-glycolic Acid) Films. J Am Chem Soc. 2006;128:5384–90.
Na D, Murty S, Lee K, Thanoo B, DeLuca P. Preparation and stability of poly(ethylene glycol) (PEG)ylated octreotide for application to microsphere delivery. AAPS Pharm Sci Tech. 2003;4:574–80.
Sophocleous AM, Zhang Y, Schwendeman SP. A new class of inhibitors of peptide sorption and acylation in PLGA. J Control Release. 2009;137:179–84.
Na DH, DeLuca PP. PEGylation of octreotide: I. Separation of positional isomers and stability against acylation by poly(D, L-lactide-co-glycolide). Pharm Res. 2005;22:736–42.
Lucke A, Göpferich A. Acylation of peptides by lactic acid solutions. Eur J Pharm Biopharm. 2003;55:27–33.
Glowka E, Sapin-Minet A, Leroy P, Lulek J, Maincent P. Preparation and in vitro-in vivo evaluation of salmon calcitonin-loaded polymeric nanoparticles. J Microencapsul. 2010;27:25–36.
Leemhuis M, van Nostrum CF, Kruijtzer JAW, Zhong ZY, ten Breteler MR, Dijkstra PJ, et al. Functionalized poly(alpha-hydroxy acid)s via ring-opening polymerization: toward hydrophilic polyesters with pendant hydroxyl groups. Macromolecules. 2006;39:3500–8.
Leemhuis M, Kruijtzer JAW, van Nostrum CF, Hennink WE. In vitro hydrolytic degradation of hydroxyl-functionalized poly(alpha-hydroxy acid)s. Biomacromolecules. 2007;8:2943–9.
Ghassemi AH, van Steenbergen MJ, Talsma H, van Nostrum CF, Jiskoot W, Crommelin DJA, et al. Preparation and characterization of protein loaded microspheres based on a hydroxylated aliphatic polyester, poly(lactic-co-hydroxymethyl glycolic acid). J Control Release. 2009;138:57–63.
Ghassemi AH, van Steenbergen MJ, Talsma H, van Nostrum CF, Crommelin DJA, Hennink WE. Hydrophilic polyester microspheres: effect of molecular weight and copolymer composition on release of BSA. Pharm Res. 2010;27:2008–17.
De Martino MC, Hofland LJ, Lamberts SW. Somatostatin and somatostatin receptors: from basic concepts to clinical applications. Prog Brain Res. 2010;182:255–80.
Feelders RA, Hofland LJ, Van Aken MO, Neggers SJ, Lamberts SWJ, De Herder WW, et al. Medical therapy of acromegaly: efficacy and safety of somatostatin analogues. Drugs. 2009;69:2207–26.
Kutz K, Nüesch E, Rosenthaler J. Pharmacokinetics of SMS 201–995 in healthy subjects. Scand J Gastroenterol Suppl. 1986;119:65–72.
Chanson P, Timsit J, Harris A. Clinical pharmacokinetics of octreotide. Therapeutic applications in patients with pituitary tumours. Clin Pharmacokinet. 1993;25:375–91.
Hofland LJ, Lamberts SWJ, Feelders RA. Role of somatostatin receptors in normal and tumoral pituitary corticotropic cells. Neuroendocrinology. 2010;92:11–6.
Modlin IM, Latich I, Kidd M, Zikusoka M, Eick G. Therapeutic options for gastrointestinal carcinoids. Clin Gastroenterol Hepatol. 2006;4:526–47.
Murty SB, Na DH, Thanoo BC, DeLuca PP. Impurity formation studies with peptide-loaded polymeric microspheres: Part II. In vitro evaluation. Int J Pharm. 2005;297:62–72.
Na DH, Youn YS, Lee SD, Son M-W, Kim W-B, DeLuca PP, et al. Monitoring of peptide acylation inside degrading PLGA microspheres by capillary electrophoresis and MALDI-TOF mass spectrometry. J Control Release. 2003;92:291–9.
Wang J, Wang BM, Schwendeman SP. Characterization of the initial burst release of a model peptide from poly (D, L-lactide-co-glycolide) microspheres. J Control Release. 2002;82:289–307.
Wang J, Wang BM, Schwendeman SP. Mechanistic evaluation of the glucose-induced reduction in initial burst release of octreotide acetate from poly(D, L-lactide-co-glycolide) microspheres. Biomaterials. 2004;25:1919–27.
Albertsson A-C, Varma IK. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules. 2003;4:1466–86.
Mehta RC, Thanoo BC, Deluca PP. Peptide containing microspheres from low molecular weight and hydrophilic poly (d, l-lactide-co-glycolide). J Control Release. 1996;41:249–57.
Li X, Deng X, Yuan M, Xiong C, Huang Z, Zhang Y, et al. Investigation on process parameters involved in preparation of poly-D, L-lactide-poly(ethylene glycol) microspheres containing Leptospira Interrogans antigens. Int J Pharm. 1999;178:245–55.
Jiang G, Thanoo BC, DeLuca PP. Effect of osmotic pressure in the solvent extraction phase on BSA release profile from PLGA microspheres. Pharm Dev Technol. 2002;7:391–9.
Jeyanthi R, Thanoo BC, Metha RC, Deluca PP. Effect of solvent removal technique on the matrix characteristics of polylactide/glycolide microspheres for peptide delivery. J Control Release. 1996;38:235–44.
Spenlehauer G, Vert M, Benoit JP, Boddaert A. In vitro and In vivo degradation of poly(D, L lactide/glycolide) type microspheres made by solvent evaporation method. Biomaterials. 1989;10:557–63.
Lucke A, Kiermaier J, Göpferich A. Peptide acylation by poly(α-hydroxy esters). Pharm Res. 2002;19:175–81.
Zhang Y, Sophocleous A, Schwendeman S. Inhibition of peptide acylation in PLGA microspheres with water-soluble divalent cationic salts. Pharm Res. 2009;26:1986–94.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Esm 1
(DOC 228 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ghassemi, A.H., van Steenbergen, M.J., Barendregt, A. et al. Controlled Release of Octreotide and Assessment of Peptide Acylation from Poly(D,L-lactide-co-hydroxymethyl glycolide) Compared to PLGA Microspheres. Pharm Res 29, 110–120 (2012). https://doi.org/10.1007/s11095-011-0517-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-011-0517-3