
ABSTRACT
Purpose
Rho-kinase regulates activation of hepatic stellate cells (HSC) during liver fibrosis, but the ubiquitous presence of this kinase may hinder examination of its exact role and the therapeutic use of inhibitors. We therefore coupled the Rho-kinase inhibitor Y27632 to a drug carrier that binds the mannose-6-phosphate insulin-like growth factor II (M6P/IGFII)-receptor which is upregulated on activated HSC.
Methods
Y27632 was coupled to mannose-6-phosphate human serum albumin (M6PHSA), and in vitro experiments were performed on primary rat HSC. Biodistribution and effect studies were performed in an acute CCl4 model in mice.
Results
Y27-conjugate remained stable in serum, while drug was efficiently released in liver homogenates. Receptor-blocking studies revealed that it was specifically taken up through the M6P/IGFII-receptor on fibroblasts, and it inhibited expression of fibrotic markers in activated HSC. In vivo, liver drug levels were significantly higher after injection of Y27-conjugate as compared to Y27632, and the conjugate accumulated specifically in HSC. After acute CCl4-induced liver injury, Y27-conjugate reduced the local activation of HSC, whereas an equimolar dose of free drug did not.
Conclusions
We conclude that specific targeting of a Rho-kinase inhibitor to HSC leads to enhanced accumulation of the drug in HSC, reducing early fibrogenesis in the liver.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Liver fibrosis is the final stage of various forms of chronic liver diseases, such as hepatitis, metabolic disorders or those caused by drug or alcohol abuse (1). Although some of the underlying causes of liver fibrosis can be treated, e.g. antiviral treatment in the case of hepatitis, at this moment there is no clinical treatment for end-stage liver cirrhosis (2). Liver fibrosis is one of the few diseases with an ever-increasing incidence (3), which warrants further research for new strategies. Due to the increasing prevalence of obesity and diabetes, nonalcoholic steatohepatitis (NASH) will give rise to more cases of severe liver fibrosis in the coming years (4).
Hepatic stellate cells (HSC) play a key role in the pathogenesis of liver fibrosis (5,6). In normal livers, they are quiescent, vitamin A-storing cells, situated near the hepatocytes in the space of Disse. In chronic diseases, stimuli from damaged hepatocytes or other liver cells lead to the development of an activated phenotype in HSC. These cells then become highly proliferative myofibroblast-like cells, which have a migratory phenotype and synthesize large amounts of extracellular matrix proteins, such as collagens and fibronectin (5,6). Activation of HSC is thus the crucial first step in the process that leads to the production of excess of extracellular matrix and eventually to end-stage liver failure in cirrhotic patients. One of the key problems in designing effective therapies is the fact that the activation of fibroblasts is a common feature of matrix turnover and wound-healing in all tissues. Therefore, a cell-specific targeted approach of this disease is required for effective treatment.
Rho-GTPase is an important regulator of cytoskeletal organization, migration and activation of HSC and myofibroblasts (7,8). The effect is mediated through various downstream mediators, of which Rho-kinase is the most important (9–11). Inhibition of Rho-kinase by the small molecular weight inhibitor Y27632 has previously been shown to reduce fibrotic parameters in primary HSC (12–16) and in several animal models of liver fibrosis (12,17,18). Apart from these anti-fibrotic effects on myofibroblasts, Rho-kinase inhibitors have other effects when administered systemically, such as a decrease in vascular smooth muscle tone (19,20), leading to an acute drop in mean arterial pressure. This would further impair the low systemic blood pressure seen in cirrhotic patients and thus deteriorate vascular homeostasis. Also, other kinase inhibitors currently in clinical use have been shown to cause side effects during treatment, which were not found during pre-clinical and clinical trials (20). In order to avoid these adverse effects and improve the effects of the Rho-kinase inhibitor, we coupled Y27632 to stellate-cell specific carrier mannose-6-phosphate human serum albumin (M6PHSA). This carrier is specifically taken up by HSC through the mannose-6-phosphate insulin-like growth factor II (M6P/IGFII)-receptor (21), which is highly and specifically upregulated on activated HSC during liver fibrosis (22,23). This multifunctional receptor traffics between the Golgi and the endosomal-lysosomal network and also shuttles to the plasma membrane (24). After binding, the protein-receptor complex is taken up into the cell through endocytosis (21). The Y27-conjugate will therefore be targeted to the liver and taken up specifically in HSC, increasing the drug dose in the target cell, whilst lowering it in non-target cells, thus reducing the chance of side effects.
In the present study, we aimed to examine the feasibility of targeting the Rho-kinase inhibitor Y27632 to the key cells in fibrogenesis, the HSC. We prepared the construct Y27632-M6PHSA and tested it in vitro in primary HSC and in vivo in an acute mouse model of CCl4-induced liver injury, characterized by local HSC activation. Biodistribution and effect studies show that the targeted Rho-kinase inhibitor is effectively delivered to the HSC, leading to high local drug concentrations within the liver, and locally reduces fibrotic parameters in vivo.
MATERIALS AND METHODS
Materials
Y27632 was purchased from Tocris Bioscience (Bristol, UK). Primary antibodies used were mouse anti-α-smooth muscle actin and anti-desmin (Sigma, St.Louis, MO), rabbit anti-human serum albumin (Cappel, Zoetermeer, Netherlands) and goat anti-M6P/IGFII-receptor (Santa Cruz Biotechnology, Santa Cruz, CA). Species-specific HRP or AP-coupled secondary antibodies were purchased from DAKO (Glostrup, Denmark).
Synthesis of Y27632-ULS-M6PHSA
The Universal Linkage System (ULS™)—developed by Kreatech Diagnostics, Amsterdam, The Netherlands—is a platinum-based linkage technology which facilitates the coupling of molecules directly to each other through the formation of a coordinative bond. The ULS™ technology has been proven to have important applications in the areas of genomics, proteomics, diagnostics, and therapeutics. The linker was conjugated to Y27632 as previously reported (25). M6P28HSA was synthesized and characterized as described elsewhere (26). ULS-Y27632 (2.1 μmol) was subsequently reacted with M6PHSA (0.14 μmol) in 20 mM tricine buffer (pH 8.5) at 37°C, and the resulting conjugate was extensively dialyzed against PBS at 4°C and purified by size-exclusion chromatography using a Superdex 200 column on an Äkta System (GE Healthcare, Uppsala, Sweden) to isolate the monomeric form of the drug-protein conjugate.
Characterization of Y27632-ULS-M6PHSA
The amount of Y27632 coupled to M6PHSA was determined by HPLC analysis after chemically displacing the drug from the carrier by incubation with 0.5 M potassium thiocyanate (KSCN) at 80°C. M6PHSA protein concentration was determined by Lowry assay (Bio-Rad, Hercules, CA). The release of drug from the Y27632-M6PHSA conjugate was determined after incubation in PBS, serum or liver homogenate until 24 h at 37°C and after 1 to 2 freeze-thaw cycles. Extraction of the Y27632 released from the carrier was performed as reported previously (25). HPLC analysis was performed on a C18 reversed-phase SunFire column (Waters, Milford, MA) using a mobile phase of water-methanol-trifluoroacetic acid (86:14:0.1, vol/vol/vol; pH 2.0) at a flow-rate of 1 ml/min. Y27632 was detected at 270 nm and eluted after circa 6.0 min. Quantitation of the drug was performed by analyzing peak areas using calibration curves prepared in the same media.
The Y27-conjugate was further analyzed by size-exclusion chromatography and anion exchange chromatography as described before (21).
In Vitro Assays
NIH-3T3 fibroblasts were cultured in DMEM (Lonza, Verviers, Belgium) containing 10% fetal calf serum (FCS) and penicillin/streptomycin. For binding studies, cells were incubated 2 h with 0.1 mg/ml Y27-conjugate or control, where necessary, preceded by a pre-incubation with M6P/IGFII-receptor antibody for 30 min.
Primary HSC were isolated from livers of male Wistar rats (>500 g, Harlan, Netherlands) according to previously published methods (27). After isolation, HSC were cultured on plastic for 10 days until activation and then used for experiments.
Real-Time PCR
Total RNA was isolated from HSC using the Absolutely RNA Microprep Kit (Stratagene, La Jolla, CA). The amount of RNA was determined using a NanoDrop UV-detector (NanoDrop Technologies, Wilmington, DE). Synthesis of cDNA was performed using the Superscript III first-strand synthesis kit (Invitrogen, Carlsbad, CA) with random primers. All primers for real-time quantitative PCR (Supplementary Table 1) were purchased from Sigma Genosys (Haverhill, UK). Gene expression levels were measured by real-time quantitative PCR on an ABI 7900HT apparatus (Applied Biosystems, Foster City, CA) with SYBR-Green PCR Master Mix (Applied Biosystems). The formation of single products was confirmed by analyzing the dissociation step at the end of each PCR reaction. Data were analyzed using the SDS 2.3 software program (Applied Biosystems). The relative amount of product was calculated using the ΔΔCt method, normalizing for the expression of the household gene GAPDH and related to the control treatment.
Animal Experiments
All animal experiments were approved by the Animal Ethics Committee of the University of Groningen, the Netherlands. All animals were purchased from Harlan (Zeist, Netherlands) and kept at 12 h light/12 h dark cycles with ad libitum chow and water.
CCl4-Induced Acute Liver Injury
For in vivo studies in the acute (72 h) model, male C57/Bl6 mice (20–22 g) received a single intraperitoneal injection of 1 ml/kg CCl4 diluted in olive oil. Control mice received olive oil. The mice were divided into 5 groups: 1) control+vehicle (PBS), 2) CCl4+vehicle (PBS), 3) CCl4+Y27632-M6PHSA (equivalent to 1.5 mg/kg/day Y27632 and 45 mg/kg/day M6PHSA), 4) CCl4+Y27632 (1.5 mg/kg/day), 5) CCl4+M6PHSA (45 mg/kg/day). All treatment groups received two i.v. injections at 24 and 48 h after CCl4 and were sacrificed 24 h after the last injection.
For biodistribution studies mice received a single i.v. injection of Y27-conjugate or Y27632 at 24 h after CCl4 injection. At 15 min, 30 min, 60 min, 3, 6, 24 and 48 h after i.v. injection of the compounds, mice were sacrificed (two mice per time-point), and liver and blood samples were collected for determination of drug levels through HPLC. Fifty mg of liver tissue was homogenized in 1:10 (w/v) PBS, whereas plasma (40 μl) was diluted 1:5 in PBS. The samples were extracted as described above to determine the levels of free drug. For the total conjugate-associated drug levels, samples were incubated overnight with 0.5 M KSCN at 80°C and then extracted as described above.
Immunohistochemistry
Immunohistochemistry was performed on 4 μm cryostat or paraffin sections. Periodic acid-Schiff’s (PAS) staining was performed on 4 μm paraffin sections. Stainings were visualized using 3, 3′-diamino-benzidine tetrahydrochloride or 3-amino-9-ethylcarbazole. Nuclei were counterstained with Mayer’s hematoxylin. Immunohistochemical stainings were quantitated using the Cell D computer program (Olympus, Hamburg, Germany).
Statistical Analysis
Results are expressed as the mean ± SD, unless otherwise specified. Statistical analyses were performed using Student’s t test or one-way ANOVA with post-hoc Bonferroni test. p < 0.05 was considered as the minimum level of significance.
RESULTS
Synthesis and Characteristics of Y27-Conjugate
To create an HSC-specific conjugate, we coupled the Rho-kinase inhibitor Y27632 to M6PHSA using the Universal Linker System (ULS™). M6PHSA was previously found to bind specifically to the M6P/IGFII-receptor which is abundantly present on activated HSC (21). The applied ULS-based method allows coupling of the aromatic nitrogen with sp2 hybridization in the pyridine ring, a common functional group in many kinase inhibitors (28), to the histidine and methionine groups of the protein (Fig. 1a). Subsequently, the conjugate was characterized using anion exchange chromatography. Introduction of Y27-ULS groups shifted the peak left compared to M6PHSA (Fig. 1b), due to addition of positive charge provided by the amine group in Y27632, confirming the coupling of Y27-ULS to the carrier. However, the resulting conjugate still retains an overall negative charge, since the Y27-ULS groups do not fully neutralize the negative charge introduced by 28 M6P groups. Size-exclusion chromatography showed that after purification approximately 90% of the Y27-conjugate was in the monomeric form (Fig. 1c).

Characteristics of the Y27632-ULS-M6PHSA conjugate. (a) Structure and diagram of the synthesis of the Y27632-ULS-mannose-6-phosphate conjugate. (b) Results of the anion exchange chromatography of HSA, M6PHSA and Y27-conjugate. (c) Results of size exclusion chromatography of Y27-conjugate. (d) Y27632 release from the conjugate at different time-points up to 24 h in PBS, liver homogenate and serum at 37°C. Collectively the data show that the Y27-conjugate has a reduced negative charge and increased molecular weight compared to M6PHSA. Significant release of drug from the carrier only occurs in liver homogenates.
The average molar coupling ratio of drug to M6PHSA was 7:1. HPLC analyses showed that there was no free drug present in the final product. We subsequently analyzed the drug release properties of the conjugate in biological media and found that only 2% or 6%, respectively, of Y27632 was released in physiological buffer and in serum at 37°C within 24 h. In contrast, in liver homogenates approximately 40% of the coupled Y27632 was released (Fig. 1d), demonstrating that Y27632 can be released from the carrier within the liver, without premature release in serum.
Binding of the conjugate to the target receptor was examined in 3T3 fibroblasts, which express the M6P/IGFII receptor (Fig. 2a). Using anti-HSA staining, we found that Y27-conjugate bound to the cells (Fig. 2b), while unmodified HSA did not bind (26). Importantly, the binding was strongly inhibited by pre-incubating the cells with an M6P/IGFII-receptor specific antibody (Fig. 2b). These data demonstrate that the conjugate binds specifically to the designated target receptor.

Binding of Y27-conjugate to M6P/IGFII-receptor. (a) Expression of the M6P/IGFII-receptor on NIH-3T3 fibroblasts. Left: control, right: M6P/IGFII-receptor staining. Original magnification 20×. (b) HSA staining showing the binding of Y27-conjugate to fibroblasts: control cells (left), conjugate-incubated cells (middle), and conjugate-incubated cells pretreated with a M6P/IGFII receptor-specific antibody (right). Note that blocking of the receptor with an antibody reduces the binding of the conjugate to the cells. Original magnification 20×.
Y27-Conjugate Inhibits Fibrotic Parameters in Activated HSC
To assess the effects of free Y27632 and Y27-conjugate on activated HSC, we analyzed mRNA expression for α-smooth muscle actin (α-SMA) in cultures of primary isolated HSC. α-SMA is the key marker reflecting the transformation of quiescent HSC into an activated, contractile phenotype (29), which starts to occur spontaneously in primary HSC after at least three days of cultivation in vitro. Additionally, we analyzed TIMP-1, which is also upregulated in activated HSC and inhibits breakdown of extracellular matrix (30). We found that the Y27-conjugate significantly reduced the expression of α-SMA and TIMP-1 (Fig. 3) in activated HSC. Effects of the targeted Rho-kinase inhibitor were similar to the effects of the untargeted inhibitor. The effects were not due to cell death, as there was no effect on cell viability, as measured by alamar blue conversion through mitochondrial activity (results not shown).

Y27-conjugate inhibits activation of hepatic stellate cells. Effect of Y27-conjugate on the mRNA levels of fibrotic markers α-SMA and TIMP-1 in isolated rat HSC after 48 h incubation with 10 μM of Y27-conjugate or equivalent amounts of Y27632 or carrier. * p < 0.05 vs. control, ** p < 0.01 vs. control, *** p < 0.001 vs. control by Student’s t-test.
In Vivo Biodistribution of Y27-Conjugate
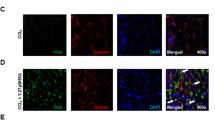
The biodistribution of the targeted conjugate was compared to that of the free drug in animals with CCl4-induced liver fibrosis 60 min after i.v. injection of the conjugate. In these livers M6P/IGFII receptor expression was already upregulated compared to control animals (Fig. 4a). No positive staining for M6P/IGFII receptor could be detected in other organs (spleen, heart, kidney or lung), neither in control nor CCl4-treated animals. The conjugate was detected using anti-HSA staining, which was found only in fibrotic livers treated with Y27-conjugate, whereas at the same time-point, no staining could be detected in the heart, kidney or lung, and only weak staining was present in the spleen (Fig. 4a). Administration of normal unmodified HSA did not result in any organ-specific staining (data not shown). Double-staining with HSC-marker desmin within the liver showed that the intrahepatic staining for the conjugate (blue) co-localized with staining for desmin (red) (Fig. 4b), confirming uptake in the target cells within the liver.

Intrahepatic localization of Y27-conjugate. (a) Upper panel: Representative photomicrographs of M6P/IGFII-receptor staining (red) in spleen, heart, kidney, lung and liver of control and CCl4-treated mice. Lower panel: Representative photomicrographs of HSA staining (red) in spleen, heart, kidney, lung and liver of CCl4-mice treated with PBS (control) and Y27-conjugate 1 h before sacrifice. Original magnification 20×. (b) Y27-conjugate localization in liver as determined by double staining for HSA (blue) and the HSC marker desmin (red) in mouse livers 1 h after injection of Y27-conjugate in CCl4-treated mice. Original magnification 40×.
Drug levels were also measured within these livers using an HPLC method. Low levels (1–2 μg/g tissue) of free drug could be detected in the livers only up to 3 h after i.v. injection of free Y27632 (Fig. 5a). In contrast, after injection of an equimolar dose of the conjugate, significantly higher levels (up to 15 μg/g tissue) of carrier-bound drug were found in the livers up to 48 h. We also determined the amount of free drug released from the conjugate in the livers after injection of the conjugate and found a slow and prolonged local release of the free drug at least up to 48 h after a single injection (Fig. 5a).

Drug concentrations in vivo and release of Y27632 from the carrier. Drug concentrations in livers after i.v. administration of Y27632 or Y27-conjugate to CCl4-mice. (a) HPLC analysis of liver extracts of CCl4-treated mice showing the biodistribution of Y27-conjugate vs. free Y27632 in livers up to 48 h after injection. (b) Accumulation of Y27-conjugate in livers of CCl4-treated mice, 10 min after injection (1 injection) and after 3 injections, 48 h, 24 h and 10 min before sacrifice (3 injections). (c) HPLC analysis of plasma of CCl4-treated mice showing the biodistribution of Y27-conjugate vs. free Y27632 in plasma up to 6 h after i.v. injection. Note that free Y27632 is only found in plasma at t = 15 min in low amounts.
Daily dosing led to a build-up of the drug concentration in fibrotic livers: while one injection led to a drug concentration of 10 μg/g liver, three injections with 24 h time intervals led to a concentration of 25 μg/g liver (Fig. 5b). These data indicate that Y27-conjugate remains in the liver after injection for more than 24 h and that free drug is slowly released, in accordance with the data presented in Fig. 5a.
In addition to liver biodistribution, we also examined distribution of the Y27-conjugate in plasma. The conjugate-bound drug could be detected in plasma up to 3 h after injection, whereas the free drug could only be detected up to 15 min after injection (Fig. 5c).
The Y27-Conjugate Reduces Rho-kinase Signaling and HSC Activation In Vivo
Livers of CCl4-treated mice showed a strong, localized upregulation of α-SMA in the central areas damaged by CCl4, representing activated, pro-fibrotic HSC. Treatment with Y27-conjugate significantly reduced α-SMA staining, indicating inhibition of HSC activation (Fig. 6a), while untargeted Y27632 did not inhibit α-SMA staining in these areas at all (Fig. 6b). The Y27-conjugate did not affect the total number of HSC in the livers, as the amount of desmin staining, reflecting both activated and unactivated HSC, was not changed significantly after treatment (Fig. 6c). These data show that the Y27-conjugate accumulated in activated HSC, thereby reducing their activity but not their number.

Y27-conjugate reduces HSC activation in acute CCl4-induced liver injury. (a) Representative pictures of immunohistochemical staining for α-SMA on liver sections of mice, 72 h after one injection of CCl4 (or olive oil in control) and treated with vehicle (PBS), Y27-conjugate, Y27632 or carrier at t = 24 and 48 h. (b) Quantitation of the area stained positive for α-SMA relative to the area stained positive in control CCl4 sections. Stainings were quantitated using the Cell D computer program, calculating the total stained area in 18–24 fields per section at 20× magnification as a percentage of the stained area in the control CCl4 sections. Data shown are the mean of 6 animals per group. * p < 0.05 vs. CCl4-PBS by one way ANOVA with Bonferroni post-hoc test. (c) Quantitation of the relative area stained positive for desmin. Stainings were quantitated at 10× magnification using the Cell D computer program as described above. Data shown are the mean of 6 animals per group.
DISCUSSION
In this study, we have shown that HSC-targeted delivery of a Rho-kinase inhibitor is possible: Y27632 coupled to M6PHSA strongly accumulated in the liver, and immunohistochemical studies showed uptake of the construct in particular in the desmin-positive cells, reflecting accumulation in HSC. Y27632 was not released in serum, so staining for the conjugate in desmin-positive HSC, 1 h after its administration, reflects uptake of the drug in this cell type.
Rho-kinase inhibitors have been used before in rat models of liver fibrosis, and these studies have shown anti-fibrotic effects of the inhibitors (12,17,18). However, in these studies, doses of 30 mg/kg of drugs were routinely used. Cell-specific delivery of Rho-kinase inhibitor may lead to a lower dose regimen. Indeed, we now have used doses of 1.5 mg/kg drug in this study and established strong pharmacological effects. We are thus able to reduce the dose needed to obtain a significant effect, while this lower dose is specifically delivered to the target cells, increasing the concentration in the target cell compared to the concentration reached after systemic administration of free drug. This results in a two-pronged approach to avoid off-target effects and increase anti-fibrotic effects of a Rho-kinase inhibitor in the liver. Avoiding adverse effects is relevant for the treatment of patients, since the Rho-kinase pathway is found to play an important role in vascular homeostasis and is deregulated in cirrhotic patients (31,32).
The Rho-kinase inhibitor was coupled to the targeted carrier system using ULS™ conjugation technology. This coupling method is based on coordination chemistry (33) and offers a rapid method for the coupling of proteins to drugs. It has been used in the past for the coupling of various kinase inhibitors to protein carriers (33–35), in particular when reactive groups in the drug are not available for conventional methods of conjugation. In addition, it provides a slow intracellular release of the drug in its active form from the carrier molecule during several days (34). This slow drug-release profile is particularly relevant for the treatment of chronic diseases such as liver fibrosis.
In vitro, the Y27632–M6PHSA conjugate was taken up by cells via the M6P/IGFII-receptor, as indicated by blockade of its uptake by IGFII-receptor-specific antibodies. Coupling of the drug to the carrier does not affect interaction with its target receptor. The drug was released from the construct in a pharmacologically active form within the target cells as reflected by a significant reduction of HSC activation in vitro. We examined the effects of drugs and constructs in cultures of primary HSC in order to be able to examine the effect of Y27632 on spontaneous HSC activation. Primary, spontaneously activated HSC are an established in vitro model for the behavior of HSC in early fibrosis (36). Both the conjugate and the free drug significantly inhibited the expression of the activation markers α-SMA and TIMP-1 in primary HSC.
In vivo as well, the Y27-conjugate attenuated α-SMA expression, confirming that Rho-kinase activity is involved in the transformation of HSC from a quiescent into an activated phenotype with contractile capabilities. Reduction of α-SMA by the Rho-kinase inhibitor is well-known (12,17,18) and might also be achieved via a direct effect of Rho-kinase on the α-SMA promotor (37). Neither the Y27-conjugate nor the free drug had any significant effect on the total number of HSC in the livers of CCl4-treated mice. In the past, several groups have looked into the effect of Rho-kinase inhibition on apoptosis and proliferation of fibroblasts. Different studies found either a pro-apoptotic effect or no effect at all (15,38,39). We have also examined this effect of the Rho-kinase inhibitor. In this study, neither the free drug nor the conjugate affected HSC viability in vitro (data not shown) or in vivo, measured as the amount of desmin-positive cells.
Importantly, administration of the HSC-targeted Rho-kinase inhibitor led to a stronger reduction of HSC activation, as reflected by a reduced α-SMA staining, than systemic treatment with the free Rho-kinase inhibitor. This shows the strength of our targeted approach, in accordance with previous papers examining cell-specific inhibition of fibrosis-related pathways in HSC using other drugs (35,40,41). Our biodistribution studies, showing elevated levels of the drug within the liver after treatment with the conjugate and a prolonged residence time of conjugate-associated drug, also are consistent with these stronger effects in the liver.
Drug concentrations in whole livers of mice receiving the targeted Rho-kinase inhibitor were much higher than in mice receiving the free drug, and concentrations remained high for a prolonged period of time. Based on the immunohistochemical data, it can be deduced that these high drug levels were concentrated in HSC, whereas the low liver uptake of free Y27632 reflects uptake by different cell-types, most likely the hepatocyte, since these cells are equipped to take up xenobiotics. Earlier studies from our group have shown that M6PHSA is rapidly endocytosed and degraded by the target cells, thus favoring intracellular drug release (26). In previous studies using this combination of carrier and linker in vivo (40,41), we also found a pharmacokinetic profile characterized by high uptake of the conjugate in the liver and a slow release of free drug. Both distribution and degree of uptake of M6PHSA found in the present study were similar to these previous chronic studies.
In conclusion, we report here the cell-specific delivery of a conjugate containing a Rho-kinase inhibitor to the M6P/IGFII-receptor on activated HSC. A cell-specific drug-conjugate needs to be evaluated on several important aspects. Accordingly, we present here data on the stability of the conjugate in plasma, release of the pharmacologically active compound in target cells and receptor-specific uptake of the conjugate. In vivo we found a pharmacokinetic profile with prolonged high drug levels in liver and localization of the conjugate within HSC, the key cells during fibrogenesis. The targeted Rho-kinase inhibitor reduced fibrotic parameters in vivo more effectively than the free drug. Our study shows the feasibility of a cell-specific delivery of kinase inhibitors, which represent an important group of new chemical entities for various serious diseases, including liver fibrosis.
Abbreviations
- α-SMA:
-
α-smooth muscle actin
- HSC:
-
Hepatic stellate cells
- M6PHSA:
-
Mannose-6-phosphate human serum albumin
- M6P/IGFII-receptor:
-
Mannose-6-phosphate/insulin-like growth factor II receptor
- ULS™:
-
Universal Linkage System
REFERENCES
Schuppan D, Afdhal N. Liver cirrhosis. Lancet. 2008;371(9615):838–51.
Thompson A, Patel K. Antifibrotic therapies: will we ever get there? Curr Gastroenterol Rep. 2010;12(1):23–9.
Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12(4):733–46. vii.
Schuppan D, Gorrell M, Klein T, Mark M, Afdhal N. The challenge of developing novel pharmacological therapies for non-alcoholic steatohepatitis. Liver Int. 2010;30(6):795–808.
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–72.
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–69.
Yee HF. Rho directs activation-associated changes in rat hepatic stellate cell morphology via regulation of the actin cytoskeleton. Hepatology. 1998;28(3):843–50.
Charest PG, Firtel RA. Big roles for small GTPases in the control of directed cell movement. Biochem J. 2007;401(2):377–90.
Wettchureck N, Offermanns S. Rho/Rho-kinase mediated signaling in physiology and pathophysiology. J Mol Med. 2002;80(10):629–38. 2007/06.
Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2004;4(6):446–56.
Kitamura K, Tada S, Nakamoto N, Toda K, Horikawa H, Kurita S, et al. Rho/Rho kinase is a key enzyme system involved in the angiotenin II signaling pathway of liver fibrosis and steatosis. J Gastroenterol Hepatol. 2007;22(11):2022–33.
Murata T, Arii S, Nakamura T, Mori A, Kaido T, Furuyama H, et al. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. J Hepatol. 2001;35(4):474–81.
Mizunuma K, Ohdan H, Fudaba Y, Tashiro H, Asahara T. ROCK inhibitor, Y-27632, inhibits anoxia-reoxygenation-induced contraction of hepatic stellate cells. Transplant Proc. 2003;35(1):111–3.
Tangkijvanich P, Melton AC, Santiskulvong C, Yee HF. Rho and p38 MAP kinase signaling pathways mediate LPA-stimulated hepatic myofibroblast migration. J Biomed Sci. 2003;10(3):352–8.
Iwamoto H, Nakamuta M, Tada S, Sugimoto R, Enjoji M, Nawata H. A p160ROCK-specific inhibitor, Y-27632, attenuates rat hepatic stellate cell growth. J Hepatol. 2000;32(5):762–70.
Fukushima M, Nakamuta M, Kohjima M, Kotoh K, Enjoji M, Kobayashi N, et al. Fasudil hydrochloride hydrate, a Rho-kinase (ROCK) inhibitor, suppresses collagen production and enhances collagenase activity in hepatic stellate cells. Liver Int. 2005;25(4):829–38.
Murata T, Arii S, Mori A, Imamura M. Therapeutic significance of Y-27632, a Rho-kinase inhibitor, on the established liver fibrosis. J Surg Res. 2003;114(1):64–71.
Tada S, Iwamoto H, Nakamuta M, Sugimoto R, Enjoji M, Nakashima Y, et al. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in rats. J Hepatol. 2001;34(4):529–36.
Hennenberg M, Biecker E, Trebicka J, Jochem K, Zhou Q, Schmidt M, et al. Defective RhoA/Rho-kinase signaling contributes to vascular hypocontractility and vasodilation in cirrhotic rats. Gastroenterology. 2006;130(3):838–54.
Force T, Krause DS, van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7(5):332–44.
Beljaars L, Molema G, Weert B, Bonnema H, Olinga P, Groothuis GM, et al. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology. 1999;29(5):1486–93.
de Bleser P, Jannes P, van Buul-Offers SC, Hoogerbrugge CM, van Schravendijk CFH, Niki T, et al. Insulin like Growth Factor-II/Mannose 6-Phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transfroming growth factor-ß in cocultures with sinusoidal endothelial cells. Hepatology. 1995;21:1429–37.
Greupink R, Bakker HI, van Goor H, de Borst MH, Beljaars L, Poelstra K. Mannose-6-phosphate/insulin-Like growth factor-II receptors may represent a target for the selective delivery of mycophenolic acid to fibrogenic cells. Pharm Res. 2006;23(8):1827–34.
Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202–12.
Prakash J, de Borst MH, Lacombe M, Opdam F, Klok PA, van Goor H, et al. Inhibition of renal rho kinase attenuates ischemia/reperfusion-induced injury. J Am Soc Nephrol. 2008;19:2086–97.
Beljaars L, Olinga P, Molema G, de Bleser P, Geerts A, Groothuis GM, et al. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver. 2001;21:320–8.
Geerts A, Niki T, Hellemans K, De Craemer D, Van Den Berg K, Lazou J, et al. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatology. 1998;27(2):590–8.
Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 2009;42(1):1–40.
Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Sem Liver Dis. 2001;21(3):311–34.
Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Sem Liver Dis. 2001;21(3):373–84.
Zhou Q, Hennenberg M, Trebicka J, Jochem K, Leifeld L, Biecker E, et al. Intrahepatic upregulation of RhoA and Rho-kinase signalling contributes to increased hepatic vascular resistance in rats with secondary biliary cirrhosis. Gut. 2006;55(9):1296–305.
Hennenberg M, Trebicka J, Biecker E, Schepke M, Sauerbruch T, Heller J. Vascular dysfunction in human and rat cirrhosis: role of receptor-desensitizing and calcium-sensitizing proteins. Hepatology. 2007;45(2):495–506.
Temming K, Fretz MM, Kok RJ. Organ and cell-type specific delivery of kinase inhibitors: a novel approach in the development of targeted drugs. Curr Mol Pharmacol. 2008;1(1):1–12.
Prakash J, Sandovici M, Saluja V, Lacombe M, Schaapveld RQ, de Borst MH, et al. Intracellular delivery of the p38 mitogen-activated protein kinase inhibitor SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1 H-imidazole] in renal tubular cells: a novel strategy to treat renal fibrosis. J Pharmacol Exp Ther. 2006;319(1):8–19.
Gonzalo T, Beljaars L, van de Bovenkamp M, Temming K, van Loenen AM, Reker-Smit C, et al. Local inhibition of liver fibrosis by specific delivery of a platelet-derived growth factor kinase inhibitor to hepatic stellate cells. J Pharmacol Exp Ther. 2007;321(3):856–65.
Iredale J. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117(3):539–48.
Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276(1):341–7.
Ikeda H, Nagashima K, Yanase M, Tomiya T, Arai M, Inoue Y, et al. Involvement of Rho/Rho kinase pathway in regulation of apoptosis in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2003;285(5):G880–6.
Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20:242–8.
Gonzalo T, Talman EG, van de Ven A, Temming K, Greupink R, Beljaars L, et al. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J Control Release. 2006;111(1–2):193–203.
Moreno M, Gonzalo T, Kok RJ, Sancho-Bru P, van Beuge M, Swart J, et al. Reduction of advanced liver fibrosis by short-term targeted delivery of an angiotensin receptor blocker to hepatic stellate cells in rats. Hepatology. 2010;51(3):942–52.
ACKNOWLEDGMENTS
This work was made possible by a grant from the European Framework program FP6 (LSHB-CT-2007-036644).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Primer sequences (DOC 25 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Beuge, M.M., Prakash, J., Lacombe, M. et al. Increased Liver Uptake and Reduced Hepatic Stellate Cell Activation with a Cell-Specific Conjugate of the Rho-kinase Inhibitor Y27632. Pharm Res 28, 2045–2054 (2011). https://doi.org/10.1007/s11095-011-0430-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-011-0430-9