
Abstract
Purpose
To obtain mathematical solutions that correlate drug and metabolite exposure and systemic bioavailability (F sys) with physiological determinants, transporters and enzymes.
Methods
A series of physiologically-based pharmacokinetic (PBPK) models that included renal excretion and sequential metabolism within the intestine and/or liver as metabolite formation organs were developed. The area under the curve for drug (AUC) and formed metabolite (AUC{mi,P}) were solved by matrix inversion.
Results
The PBPK models revealed that AUC{mi,P} was dependent on dispositional parameters (transport and elimination) for the drug and metabolite. The solution was unique for each metabolite formation organ and was dependent on the type of drug and metabolite elimination organs. The AUC ratio of the formed metabolite after oral and intravenous drug dosing was useful for determination of the fraction absorbed (F abs) and not the systemic bioavailability (F sys) when either intestine or liver was the only drug elimination organ.
Conclusions
The AUC ratio of the formed metabolite after oral and intravenous drug dosing differed from that for drug and would not provide F sys. However, the AUC ratio of the formed metabolite for oral and intravenous drug dosing furnished the estimate of F abs when intestine or liver was the only drug metabolic organ.



Similar content being viewed by others


References
Löbenberg R, Amidon GL. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm. 2000;50:3–12.
Rowland M. Infuence of route of administration on drug bioavailability. J Pharm Sci. 1972;61:70–4.
Wagner JG. An overview of the analysis and interpretation of bioavailability studies in man. Pharmacology. 1972;8:102–17.
Koch-Weser J. Bioavailability of drugs (first of two parts). N Engl J Med. 1974;291:233–7.
Koch-Weser J. Bioavailability of drugs (second of two parts). N Engl J Med. 1974;291:503–6.
Riegelman S, Rowland M. Effect of route of administration on drug disposition. J Pharmacokinet Biopharm. 1973;1:419–34.
Rescigno A. Area under the curve and bioavailability. Pharmacol Res. 2000;42:539–40.
Cornaire G, Woodley J, Hermann P, Cloarec A, Arellano C, Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm. 2004;278:119–31.
Batrakova E, Lee S, Li S, Venne A, Alakhov V, Kabanov A. Fundamental relationships between the composition of pluronic block copolymers and their hypersensitization effect in MDR cancer cells. Pharm Res. 1999;16:1373–9.
Minko T, Batrakova EV, Li S, Li Y, Pakunlu RI, Alakhov VY, et al. Pluronic block copolymers alter apoptotic signal transduction of doxorubicin in drug-resistant cancer cells. J Control Release. 2005;105:269–78.
Johnson BM, Charman WN, Porter CJ. An in vitro examination of the impact of polyethylene glycol 400, Pluronic P85, and vitamin E D-alpha-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSci. 2002;4:E40.
Kabanov AV, Batrakova EV, Alakhov VY. An essential relationship between ATP depletion and chemosensitizing activity of Pluronic block copolymers. J Control Release. 2003;91:75–83.
Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J Pharm Sci. 2002;91:1991–2002.
Shono Y, Nishihara H, Matsuda Y, Furukawa S, Okada N, Fujita T, et al. Modulation of intestinal P-glycoprotein function by cremophor EL and other surfactants by an in vitro diffusion chamber method using the isolated rat intestinal membranes. J Pharm Sci. 2004;93:877–85.
Zhang H, Yao M, Morrison RA, Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res. 2003;26:768–72.
Cornaire G, Woodley JF, Saivin S, Legendre JY, Decourt S, Cloarec A, et al. Effect of polyoxyl 35 castor oil and Polysorbate 80 on the intestinal absorption of digoxin in vitro. Arzneimittelforschung. 2000;50:576–9.
Risovic V, Sachs-Barrable K, Boyd M, Wasan KM. Potential mechanisms by which Peceol increases the gastrointestinal absorption of amphotericin B. Drug Dev Ind Pharm. 2004;30:767–74.
Hugger ED, Audus KL, Borchardt RT. Effects of poly(ethylene glycol) on efflux transporter activity in Caco-2 cell monolayers. J Pharm Sci. 2002;91:1980–90.
Collnot EM, Baldes C, Wempe MF, Hyatt J, Navarro L, Edgar KJ, et al. Influence of vitamin E TPGS poly(ethylene glycol) chain length on apical efflux transporters in Caco-2 cell monolayers. J Control Release. 2006;111:35–40.
Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–46.
Sancho-Chust V, Fabra-Campos S, Gomez-Meseguer V, Bengochea M, Martin-Villodre A. Experimental studies on the influence of surfactants on intestinal absorption of drugs. Cefadroxil as model drug and sodium lauryl sulfate as model surfactant: studies in rat colon. Arzneimittelforschung. 1995;45:595–601.
Kim IW, Yoo HJ, Song IS, Chung YB, Moon DC, Chung SJ, et al. Effect of excipients on the stability and transport of recombinant human epidermal growth factor (rhEGF) across Caco-2 cell monolayers. Arch Pharm Res. 2003;26:330–7.
Kotze AF, Luessen HL, de Leeuw BJ, de Boer BG, Verhoef JC, Junginger HE. N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: in vitro evaluation in intestinal epithelial cells (Caco-2). Pharm Res. 1997;14:1197–202.
van der Merwe SM, Verhoef JC, Verheijden JH, Kotze AF, Junginger HE. Trimethylated chitosan as polymeric absorption enhancer for improved peroral delivery of peptide drugs. Eur J Pharm Biopharm. 2004;58:225–35.
Bittner B, Bravo González RC, Walter I, Kappas M, Huwyler J. Impact of Solutol HS 15 on the pharmacokinetic behaviour of colchicine upon intravenous administration to male Wistar rats. Biopharm Drug Dispos. 2004;25:37–49.
Smith NF, Acharva MR, Desai N, Figg WD, Sparreboom A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol Ther. 2005;4:815–8.
Bravo González RC, Huwyler J, Boess F, Walter I, Bittner B. In vitro investigation on the impact of the surface-active excipients Cremophor EL, Tween 80 and Solutol HS 15 on the metabolism of midazolam. Biopharm Drug Dispos. 2004;25:37–49.
Ren X, Mao X, Si L, Cao L, Xiong H, Qiu J, et al. Pharmaceutical excipients inhibit cytochrome P450 activity in cell free systems after systemic administration. Eur J Pharm Biopharm. 2008;70:279–88.
FDA Guidance for Industry. Bioavailability and bioequivalence studies for orally administered drug products—general considerations. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), March 2003.
Rosenblum SE, Lam J. Bioequivalence parameters of parent drug and its first-pass metabolite: comparative sensitivity to sources of pharmacokinetic variability. Drug Dev Ind Pharm. 1997;23:237–344.
Jackson AJ. The role of metabolites in bioequivalency assessment. III. Highly variable drugs with linear kinetics and first pass effect. Pharm Res. 2000;17:1432–6.
Tucker G, Rostami A, Jackson P. Metabolite measurements in bioequivalence studies. Theoretical considerations. In: Midha KK, Blume HH, editors. Bio-International: bioavailability, bioequivalence and pharmacokinetics. Stuttgart: Medpharm Scientific Publishers; 1992. p. 163–70.
Midha KK, Rawson MJ, Hubbard JW. Commentary. The role of metabolites in bioequivalence. Pharm Res. 2004;21:1331–4.
Pang KS, Morris ME, Sun H. Formed and preformed metabolite: Facts and comparisons. J Pharm Pharmacol. 2008;60:1247–75.
Chen M-L, Jackson AJ. The role of metabolites in bioequivalency assessment. I. linear pharmacokinetics without first pass effect. Pharm Res. 1991;8:25–32.
Chen M-L, Jackson AJ. The role of metabolites in bioequivalency assessment. II. Drugs with linear pharmacokinetics and first-pass effect. Pharm Res. 1995;12:700–8.
Sun H, Pang KS. Disparity in intestine disposition between formed and preformed metabolites and implications: A theoretical study. Drug Metab Dispos. 2009;37:187–202.
Pang KS. Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review Series). Drug Metab Dispos. 2003;31:1507–19.
Kwan KC. Oral bioavailability and first-pass effects. Drug Metab Dispos. 1997;25:1329–35.
Yamazaki S, Toth LN, Kimoto E, Bower J, Skaptason J, Romero D, et al. Application of stable isotope methodology in the evaluation of the pharmacokinetics of (S, S)-3-(3-(methylsulfonyl)phenyl)-1-propylpiperidine hydrochloride in rats. Drug Metab Dispos. 2009;37:937–45.
Peters SA. Identification of intestinal loss of a drug through physiologically based pharmacokinetic simulation of plasma concentration-time profiles. Clin Pharmacokinet. 2008;47:245–59.
Pang KS, Gillette JR. Sequential first-pass elimination of a metabolite derived from a precursor. J Pharmacokinet Biopharm. 1979;7:275–90.
Weiss M. A general model of metabolite kinetics following intravenous and oral administration of the parent drug. Biopharm Drug Dispos. 1988;9:159–76.
Weiss M. Use of metabolite AUC data in bioavailability studies to discriminate between absorption and first-pass extraction. Clin Pharmacokinet. 1990;18:419–22.
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5.
Dyke TM, Hubbell JA, Sams RA, Hinchcliff KW. Hepatic blood flow in horses during the recuperative period from maximal exercise. Am J Vet Res. 1998;59:1476–80.
Shayeganpour A, Hamdy DA, Brocks DR. Pharmacokinetics of desethylamiodarone in the rat after its administration as the preformed metabolite, and after administration of amiodarone. Biopharm Drug Dispos. 2008;29:159–66.
Peyrou M, Bousquet-Melou A, Laroute V, Vrins A, Doucet MY. Enrofloxacin and marbofloxacin in horses: comparison of pharmacokinetic parameters, use of urinary and metabolite data to estimate first-pass effect and absorbed fraction. J Vet Pharmacol Ther. 2006;29:337–44.
Bennett PN, Aarons LJ, Bending MR, Steiner JA, Rowland M. Pharmacokinetics of lidocaine and its deethylated metabolite: dose and time dependency studies in man. J Pharmacokinet Biopharm. 1982;10:265–81.
Lê KT, Maurice H, du Souich P. First-pass metabolism of lidocaine in the anesthetized rabbit. Contribution of the small intestine. Drug Metab Dispos. 1996;24:711–6.
Hajda JP, Jähnchen E, Øie S, Trenk D. Sequential first-pass metabolism of nortilidine: the active metabolite of the synthetic opioid drug tilidine. J Clin Pharmacol. 2002;42:1257–61.
Lo MW, Goldberg MR, McCrea JB, Lu H, Furtek CI, Bjornsson TD. Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin Pharmacol Ther. 1995;58:641–9.
Lindberg RL, Syvälahti EK. Metabolism of nomifensine after oral and intravenous administration. Clin Pharmacol Ther. 1986;39:378–83.
Lindberg RL, Syvälahti EK, Pihlajamäki KK. Disposition of nomifensine after acute and prolonged dosing. Clin Pharmacol Ther. 1986;39:384–8.
Acknowledgment
This work was supported by the Canadian Institutes for Heath Research, MOP89850.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
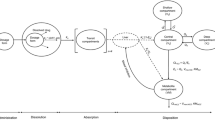
Mass balance equations and the corresponding matrices for the physiologically based pharmacokinetic model (case 1 to case 4, as shown in Figs. 1, 2 and 3)
- Q:
-
blood flow rate
- V:
-
blood or tissue volume
- P:
-
parent drug
- Mi:
-
the primary metabolite of interest
- SB:
-
systemic blood, used as subscripts
- HP:
-
highly perfused organ, used as subscript
- PP:
-
poorly perfused organ, used as subscript
- Intb:
-
intestinal blood, used as subscript
- Int:
-
intestinal tissue, used as subscript
- Lumen:
-
intestinal lumen, used as subscript
- LB:
-
liver blood, used as subscript
- L:
-
liver tissue, used as subscript
- PV:
-
portal vein, used as subscript
- HA:
-
hepatic artery, used as subscript
- CLr, CLr{mi}:
-
apparent renal clearances of the parent drug and the metabolite, Mi, respectively
- \( {{CL}}_{\rm{d1}}^{\rm{I}}{,}{\kern 1pt} {{CL}}_{\rm{d2}}^{\rm{I}}{\kern 1pt} {\kern 1pt} \) :
-
basolateral influx and efflux clearances of enterocytes, respectively
- CLint,met1,I :
-
metabolic intrinsic clearance for formation of the Mi in the intestinal tissue
- CLint,met2,I :
-
metabolic intrinsic clearance for formation of other metabolites in the intestinal tissue
- CLint,sec,I :
-
secretory intrinsic clearance for drug in the intestinal tissue
- ka :
-
rate constant of drug absorption in the intestine
- kg :
-
rate constant of intestinal transit and degradation
- \( {{CL}}_{\rm{d1}}^{\rm{H}}{,}{\kern 1pt} {{CL}}_{\rm{d2}}^{\rm{H}}{\kern 1pt} {\kern 1pt} \) :
-
basolateral influx and efflux clearances of the hepatocyte, respectively
- CLint,met1,H :
-
metabolic intrinsic clearance for formation of the metabolite of interest in liver
- CLint,met2,H :
-
metabolic intrinsic clearance for formation of other metabolites in the liver
- CLint,sec,I :
-
secretory intrinsic clearance of drug in the liver
- {mi} and {mii}:
-
symbols used to qualify the parameters for primary metabolites formed in intestine and other primary metabolite formed in liver for case 3
-
(1)
Case 1 (see Fig. 1 for the model scheme)
In systemic blood (denoted by the subscript, SB),
In highly perfused organs (denoted by the subscript, HP),
In poorly perfused organs (denoted by the subscript, PP),
In intestinal blood (denoted by the subscript, Intb),
In intestinal tissue (denoted by the subscript, Int),
In intestinal lumen (denoted by the subscript, lumen),
In liver blood (denoted by the subscript, LB),
In liver tissue (denoted by the subscript L),
-
(2)
Case 2 (see Fig. 2 for the model scheme)
In systemic blood (denoted by the subscript, SB),
In highly perfused organs (denoted by the subscript, HP),
In poorly perfused organs (denoted by the subscript, PP),
In intestinal blood (denoted by the subscript, Intb),
In intestinal tissue (denoted by the subscript, Int),
In intestinal lumen (denoted by the subscript, lumen),
In liver blood (denoted by the subscript, LB),
In liver tissue (denoted by the subscript, L),
-
(3)
Case 3 (see Fig. 3 for the model scheme, different metabolites were formed in intestine (as metabolite mi) and liver (as metabolite mii))
In systemic blood (denoted by the subscript, SB),
In highly perfused organs (denoted by the subscript, HP),
In poorly perfused organs (denoted by the subscript, PP),
In intestinal blood (denoted by the subscript, Intb),
In intestinal tissue (denoted by the subscript, Int),
In intestinal lumen (denoted by the subscript, lumen),
In liver blood (denoted by the subscript, LB),
In liver tissue (denoted by the subscript, L),
-
(4)
Case 4 (see Fig. 3 for the model scheme, the same metabolite was formed in intestine and liver)
In systemic blood (denoted by the subscript, SB),
In highly perfused organs (denoted by the subscript, HP),
In poorly perfused organs (denoted by the subscript, PP),
In intestinal blood (denoted by the subscript, Intb),
In intestinal tissue (denoted by the subscript, Int),
In intestinal lumen (denoted by the subscript, lumen),
In liver blood (denoted by the subscript, LB),
In liver tissue (denoted by the subscript, L),
Rights and permissions
About this article
Cite this article
Sun, H., Pang, K.S. Physiological Modeling to Understand the Impact of Enzymes and Transporters on Drug and Metabolite Data and Bioavailability Estimates. Pharm Res 27, 1237–1254 (2010). https://doi.org/10.1007/s11095-010-0049-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0049-2