
Abstract
Purpose
Previously, we have shown that the amphiphilic oligopeptide SA2 (Ac-Ala-Ala-Val-Val-Leu-Leu-Leu-Trp-Glu-Glu-COOH) spontaneously self-assemble into nano-sized vesicles in aqueous environment. Relative weak individual intermolecular interactions dominate such oligopeptide assemblies. In this study we aimed at improving the stability of such peptide vesicles by covalently crosslinking the oligopeptide vesicles using disulfide bonds. Two and three cysteines were introduced in the SA2 peptide sequence to allow crosslinking (Ac-Ala-Cys-Val-Cys-Leu-(Leu/Cys)-Leu-Trp-Glu-Glu-COOH).
Results
Upon disulfide formation the crosslinked vesicles remained stable under conditions that disrupted the non-crosslinked peptide vesicles. The stabilized vesicles were more closely examined in terms of particle size (distribution) using atomic force microscopy, cryogenic electron microscopy, as well as dynamic light scattering analysis, showing an average particle radius in number between 15 and 20 nm. Using entrapment of calcein it was shown that intermolecular crosslinking of peptides within the vesicles did not affect the permeability for calcein.
Conclusion
Introduction of cysteines into the hydrophobic domain of the SA2 amphiphilic oligopeptides is a feasible strategy for crosslinking the peptide vesicles. Such small crosslinked oligopeptide vesicles may hold promise for drug delivery applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Self-assembly of peptides can result in a whole array of different supramolecular morphologies (1–6). Long polypeptides as well as oligopeptides have been exploited to develop hydrogels, nanoribbons, or peptide sheets, several of these new (bio)materials displaying unique and useful macromolecular characteristics (7–9). Previously, we developed amphiphilic oligopeptides that assemble into nanosized vesicles (10). These peptides display a conical design, in which the amino acids that constitute the hydrophobic domain increase in size towards the hydrophilic part, while charged amino acids function as the hydrophilic domain (e.g. Ac-Ala-Ala-Val-Val-Leu-Leu-Leu-Trp-Glu-Glu; SA2). Nano-vesicles formed spontaneously upon hydration of these peptides and could be reversibly disrupted and re-assembled upon changes in environmental factors like pH (9).
The individual intermolecular interactions that stabilize these oligopeptide vesicles, like hydrophobic interactions, van der Waals forces, and hydrogen bonding, are relatively weak. The supramolecular structure exists in equilibrium with oligopeptides in free form, displaying a critical aggregation concentration (CAC) of around 0.5 μM for the SA2 peptide (10,11). As a consequence, the supramolecular peptide assemblies destabilize upon dilution, and for some applications of these peptide vesicles a structure that is more stable may be desired. For example, in the case of drug delivery application, injection into the bloodstream or tissue may destabilize the peptide assemblies by binding of the free peptides to blood components or by extensive dilution of the assemblies.
Hence, in this study we aimed at stabilizing the previously developed oligopeptide vesicles. We used intermolecular disulfide formation, which is controllable as well as stable in aqueous environment, introducing two and three cysteines into the hydrophobic domain of the oligopeptide sequence.
MATERIALS & METHODS
Materials
Chemicals were from Sigma (St. Louis, USA) unless indicated otherwise. Hepes, acetic anhydride was from Fluka, Ellman’s reagent was from Pierce (Rockford, USA). pET SUMO and chemically competent TOP10 and BL21(DE3) E. coli cells were from Invitrogen (Carlsbad, USA). SUMO protease was from (LifeSensors, Malvern, USA).
DNA Design and Construction
Two sets of complementary oligo DNA (5′ gcgtgcgtgtgtctgctgctgtgggaagaatgaggatcca 3′; 5′ ggatcctcattcttcccacagcagcagacacacgcacgca 3′ and gcgtgcgtgtgtctgtgcctgtgggaagaatgaggatcca 3′; 5′ ggatcctcattcttcccacaggcacagacacacgcacgca 3′) were designed to have 3′ A-overhangs for directional ligation (TA cloning) and a BamHI recognition site after the peptide coding region and stop codon. Both complementary oligos were annealed at a 5.0 μM DNA concentration using temperature gradients (cooling at 0.2° per second; 10 min 95°C, cooling to 30°C, 10 min 75°C, cooling to 30°C, 10 min 65°C, cooling to 30°C) and ligated into pET SUMO with T4 DNA ligase (Fermentas, Burlington, USA). After transformation of TOP10 E. Coli cells, colonies were screened for the right DNA construct by colony PCR and subsequent BamHI (Fermentas, Burlington, USA) restriction analysis on purified plasmid DNA. Correct insertion of the dsDNA was confirmed by DNA sequencing (BaseClear, Leiden, The Netherlands).
Peptide Biosynthesis
E. Coli BL21(DE3) cells were transformed with plasmids encoding the SUMO-peptide fusion protein, and expression and protein purification were performed as described before (10). Briefly, bacteria were cultured in a 5 L Luria Broth pO2-stat fed-batch fermentation. Protein expression was induced by addition of isopropyl-β-D-thiogalactopyranoside (IPTG) (Fisher Emergo, Landsmeer, The Netherlands) to a final concentration of 1.0 mM. After 3 h, cells were harvested by centrifugation for 15 min at 3,500 g and lysed by means of a single freeze-thaw step and passing twice through an Avestin C5 cell-cracker (ATA Scientific, Lucas Heights, Australia). The cleared lysate, the His-tagged fusion protein was purified on a 20 ml HisTrap column (GE Healthcare, Upsala, Sweden) by automated flow purification. After elution with a 300 mM phosphate buffer (pH 8.0) containing 400 mM imidazole and buffer exchange to Hepes buffered saline (HBS, 20 mM Hepes, 150 mM NaCl, pH 8.0) supplemented with 2.5 mM dithiotreitol (DTT), the peptides were cleaved off the fusion protein by incubation with 2 u/ml SUMO protease at 30°C. The peptide was purified from the SUMO protein and protease using a size exclusion column of Sephadex™ G–25 Fine material (GE Healthcare, Upsala, Sweden) in a buffer of 10 mM Hepes, 75 mM NaCl, 2.5 mM DTT, pH 8.0. Subsequently, N-terminally acetylation was performed for 1 h in 50% ddH2O, 25% methanol 25% acetic anhydride (v/v). Acetylation was confirmed (>95%) by determination of free amines using 2,4,6-trinitrobenzene sulfonic acid (Pierce, Rockford, USA) according to manufacturer’s protocol. The solvents were removed by reduced pressure evaporation. The correct peptide mass was confirmed by mass spectrometry after S-carboxymethylation of the cysteine residues by iodoacetic acid (12) (SA2C2 expected mass 1294.1; found mass 1294.0; SA2C3 expected mass 1282.4; found mass 1281.5 (diiodoacetylated)).
Gel Electrophoresis
For sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), samples were boiled in Laemmli sample buffer without (non-reducing conditions) or with 25% β-mercaptoethanol (v/v) for 5 min and loaded onto a 15% acrylamide gel. Electrophoresis was performed at room temperature, applying 10 mA per gel until the running front reached the end of the gel. The gel was fixed in a 38/50/12 (v/v/v) mixture of water/methanol/acetic acid followed by silver staining. PageRuler prestained protein ladders were obtained from Fermentas (Burlington, USA).
Peptide Self-assembly
The acetylated peptides were reconstituted in ddH2O and washed as follows. Typically, 8.0 μmol of peptides were sedimented after acidifying with HCl (pH < 2.5) the peptide solution (15 ml) by centrifugation for 30 min at 13,000 g, and the supernatant was carefully removed. Peptide self-assembly was performed by adding 3.0 ml of 20 mM phosphate buffer (PB), pH 8.0 on top of the pellet, excluding mechanical input. The peptide solutions were exposed to air for 16 h. The peptide concentration in the dispersions was determined by UV absorbance of the tryptophan residue at 280 nm, using a molar extinction coefficient of 5,690 l mol−1 cm−1. The measurements were checked on light scattering and, if needed corrected for according to ref. 10 (13).
Ellman’s Reaction
To the peptide dispersions diluted in 0.1 M sodium phosphate, 1.0 mM EDTA, pH 7.0 Ellman’s Reagent was added, to a concentration of 71 μg/ml. The conversion to 2-nitro-5-thiobenzoic acid was monitored at 412 nm in a Shimadzu spectrophotometer. The conversion was considered complete when the signal became stable over time (typically after 16 h). Subsequently, the free thiols were quantified using the molar extinction coefficient of 2-nitro-5-thiobenzoic acid (14 150 l mol−1 cm−1)(14).
Dynamic Light Scattering
Dynamic light scattering measurements were performed in a Malvern ALV CGS-3 goniometer (Malvern Instruments, Malvern, U.K.) containing a He-Ne laser source (λ = 632.8 nm, 22 mW output power) under an angle of 90°. The DLS time correlation was analyzed by ALV Correlator 3.0 software (ALV, Langen, Germany). The refractive indices used were 1.333 (water) and 1.431 (DMF). In general, the solvents used to dilute the peptides were filtered through a 0.2 μm filter before use, and three independent light scattering experiments were performed on the peptide samples.
Electron Microscopy
Cryogenic transmission electron microscopy (TEM) analysis was performed on 2.5 mM peptide dispersions. Samples were applied on glow-discharged 200 mesh grids, covered with Quantifoil holey carbon foil (Micro Tools GmbH, Jena, Germany) and blotted for 0.5 s at 100% relative humidity. Immediately, the samples were vitrified by plunging the grid into liquid ethane using a Vitrobot (FEI Company, Eindhoven, The Netherlands) and subsequently put in dry liquid nitrogen. Grids were introduced in a Tecnai12 transmission electron microscope (FEI co, Eindhoven, The Netherlands). Samples were observed at 120 kV with low-dose imaging conditions to avoid melting of the vitrified film. Images were recorded on TemCam-0124 camera (TVIPS GmBH, Gauting, Germany) and processed with AnalySIS software.
Atomic Force Microscopy
Freshly cleaved mica was incubated for 5 min with 0.1 mg/ml poly-ornithine (30–70 kDa, Sigma, St. Louis, USA) and subsequently washed 4 times with water. A 0.3 mM peptide dispersion was applied on the coated mica and incubated for 5 min. The peptide dispersion was removed, the mica was rinsed, and 10 mM PB was applied. Tapping mode AFM was performed in aqueous environment with a Multimode AFM and Nanoscope IIIa controller, equipped with a Silicon Nitride NP cantilever (Veeco, Santa Barbara, USA). A 12 μm piezoscanner (E scanner) was employed for imaging (Veeco, Santa Barbara, USA).
Calcein Entrapment and Release Assay
Twenty five nanomol peptide was reduced in 20 mM freshly prepared DTT for 40 min at 45°C. The peptides were precipitated by addition of 1.0 M HCl to pH < 3.0 and centrifuged for 10 min at 13,000 g, and the pellet was washed twice with 100 μl 10 mM HCl. Peptide self-assembly was performed in 50 μl calcein solution (40 mg/ml calcein, 20 mM Hepes, pH 8.0) at anaerobic conditions (degassed solution, under argon) and subsequently exposed to air for 16 h. Size exclusion spin columns were prepared as described by Fry et al. (15). Four milliliters of G25 Sephadex Medium Coarse size (GE Healthcare, Upsala, Sweden) swollen in Hepes buffered saline (HBS, 150 mM NaCl, 20 mM Hepes, pH 8.0) was used per column. Fifty microliters of the peptide dispersion in calcein solution was applied on top of the column and centrifuged for 3 min at 350 g. The flow through was collected (fraction 1), and 50 μl of isotonic HBS were used for elution of the subsequent fractions (7 fractions in total). Forty microliters of each fraction were analyzed for calcein fluorescence as previously described (10). To measure kinetics of calcein release from peptide vesicles, the void volume fractions (1,2) were directly measured over time in a Horiba Fluorolog fluorescence spectrometer (excitation 495 nm, emission 518 nm, slits 1.0 nm). The total encapsulated calcein was quantified after reduction in 20 mM DTT and disruption of the supramolecular structure in DMF (90% DMF, 10% HBS). Concentration series of calcein from 1.0 µM to 1.0 nM in 90% DMF, 10% HBS was used for calibration.
RESULTS AND DISCUSSION
Peptide Design and Production
Amphiphilic peptides that were previously shown to assemble into vesicles were used as the basic motif to develop crosslinkable peptide vesicles. The hydrophobic domain of the peptide exhibits a conical shape, in which the amino acid side groups decrease in size from the interface along its hydrophobic chain (Table I) (10). Considering that thiols can function as an elegant and reversible route to stabilize the peptide assemblies (16), two (SA2C2) or three (SA2C3) cysteines were introduced in the hydrophobic domain (Table I). The cysteine-containing peptides fused to the small-ubiquitin modifying protein (SUMO) were recombinantly produced in E. coli as previously described (10). SDS-PAGE (Fig. 1) showed that the purified SUMO-SA2C2 and SUMO-SA2C3 fusion proteins appeared around 17 kDa. Although the expected mass of the fusion proteins (15 kDa) is lower, the SUMO protein is known to run at a somewhat higher apparent molecular weight on SDS-PAGE (17). The SUMO protease specifically cleaves SUMO protein by means of recognition of the folded SUMO protein (17). In lanes 4 and 5 (Fig. 1a) an intermediate of the cleavage process (1 h incubation at 30°C) was analyzed on the gel, and the two different proteins are visible: the upper band corresponds to the SUMO-peptide fusion protein and the lower band to the cleaved SUMO protein. Because of the small size of the released peptide (MW 1.2 kDa), the monomeric peptide is not visible on gel under the tested conditions. SDS-PAGE showed that almost quantitative cleavage (>95%) was achieved by prolongation of the incubation time to 4 h at 30°C (data not shown). Since the SUMO-peptide fusion protein contains two (SUMO-SA2C2) or three (SUMO-SA2C3) thiols, protein dimerization was also investigated using SDS-PAGE under non-reducing conditions. As can be seen in Fig. 1b, the non-cleaved fusion proteins in lanes 3 (SUMO-SA2C2) and 5 (SUMO-SA2C3) revealed the presence of protein dimers, whereas after cleavage (Figs. 1b, lanes 2 and 4) dimerization of SUMO was not observed. This is expected since the SUMO protein itself does not contain any cysteines.

SDS-PAGE analysis on the purified fusion proteins. Reducing conditions (A): lane 1 SUMO-SA2C2, lane 2 SUMO-SA2C3, lane 3 molecular weight markers, lane 4 SUMO-SA2C2 partially cleaved, lane 5 SA2C3 partially cleaved. Non-reducing conditions (B): lane 1 the molecular weight marker, lane 2 SUMO-SA2C2 cleaved and not cleaved (lane 3), lane 4 SUMO-SA2C3 cleaved and not cleaved (lane 5).
Peptide Self-assembly and Intermolecular Crosslinking of the Peptide Vesicles
The self-assembling capacity of acetylated SA2C2 and SA2C3 peptides in phosphate buffer (PB) at pH 8.0 was monitored by dynamic light scattering. Table II shows that both peptides formed supramolecular structures with nanoscale dimensions in PB. Using the Ellman’s free thiol assay, formation of disulfide bonds within the peptide vesicles was confirmed (5% free thiol remained). The SA2C3 particle radius (75 nm) was in good agreement with the non-crosslinked SA2 peptide assemblies (63 nm); in the case of SA2C2 peptides a somewhat larger mean particle radius (106 nm) was observed. To investigate whether the existence of the observed assemblies relied on the oxidation of the cysteines, DTT (20 mM) was added. Subsequent DLS analysis showed that the assemblies were stable under reducing conditions, and loss of scattering intensity was not observed (Table II). Moreover, the reducing conditions did not significantly change the mean particle size, indicating that the assembled structures were not affected by reduction of disulfide bonds.
In order to assess whether this disulfide bond formation indeed stabilized the peptide assemblies, the organic solvent DMF was added to the peptide dispersion to a final concentration of 95% (v/v). This solvent disrupts hydrogen bonds as well as hydrophobic clustering effects and was found a good solvent for hydrophobic or amphiphilic (poly)peptides (18). When the peptides were reduced with DTT and subsequently injected into DMF, the particle light scattering dropped to background levels instantaneously (Table II), indicating full disintegration of the assembled structures. In contrast, when the oxidized peptide assemblies were brought into DMF, the scattering intensity remained and was stable over time (Table II). DLS analysis on the assembled structures in DMF indicated a mean particle radius of 50–65 nm. Besides direct effects, noncovalent interparticle interactions may be broken by DMF as well, which may explain the observed lower mean particle radius. In conclusion, the stability of the oxidized peptide assemblies in DMF demonstrated that inter-peptide disulfide bonds were formed, which covalently crosslinked the peptide assemblies.
The particle size derived from DLS measurements was more closely examined. In Fig. 2, the particle size distribution of SA2C2 and SA2C3 assemblies was plotted as a function of the scattering intensity. The peptide assemblies displayed a wide distribution of particle sizes. The peak around 100 nm, which was the dominant peak, confirms the mean particle radii of Table II. Besides the 100 nm particles, light scattering by smaller particles (peak around 10 nm, radius) was detected as well for the oxidized SA2C2 and SA2C3 peptide dispersions (Fig. 2a and b). Since the size distribution graphs are based on the scattering intensity, in number the smaller particles may constitute the major fraction of the peptide structures. In Fig. 2b also an ~2,000 nm peak was observed, which was not present for the SA2C2 dispersion. The 2,000 nm peak diminished when the oxidized assemblies were brought in DMF (Supplementary Information), which may indicate some clustering of particles in aqueous environment. The ~10 nm (radius) peak remained in DMF for both peptide assemblies.

Dynamic light scattering size distribution graphs of the oxidized SA2C2 (A) and SA2C3 (B) peptide assemblies in 10 mM PB of pH 8.0.
Cryogenic Electron Microscopy Analysis of Peptide Assemblies
As DLS cannot reveal anything about the morphology of the peptide assemblies, cryogenic transmission electron microscopy (cryoTEM) was performed. The vitrification of the sample in cryoTEM allowed the direct examination of the peptide structures as present in solution. Representative micrographs of the crosslinked peptide assemblies are shown in Fig. 3. As can be seen in the micrographs, both SA2C2 and SA2C3 samples (oxidized) showed spherical structures similar to what we have previously observed for SA2 peptides, which formed vesicles (10). The size of the particles was examined by measuring the diameter of 100 particles on average at random tracks. Table III shows that the mean particle radii for both the SA2C2 and SA2C3 assemblies was <20 nm, which is substantially smaller than the size detected by DLS (75 nm or larger; Table II). Some assemblies indeed were clustered, which may in part explain the larger average sizes detected with DLS (Fig. 2).

Cryogenic transmission electron microscopy micrographs of the crosslinked SA2C2 (A) and SA2C3 (B) peptide assemblies.
Atomic Force Microscopy
Atomic Force Microscopy (AFM) was used to further characterize particle morphology and size distribution of the SA2C2 and SA2C3 peptide assemblies. After coating of the mica with polyornithine (10), the negatively charged peptide assemblies were immobilized on the surface, and AFM was performed in solution. Fig. 4 shows representative AFM micrographs of the crosslinked peptide assemblies. In accordance with the cryoTEM, only spherical particles were observed with AFM. The particle size and size distribution were analyzed using AFM WSXM analysis software (19). As can be seen in Table III, the average particle sizes derived from the AFM measurements are in good agreement with the average sizes found by cryoTEM. The mean particle radius from either technique is ~20 nm for the SA2C3 assemblies, and around 16 nm for the SA2C2, respectively. When the reduced assemblies were examined with AFM, neither significant difference in particle size nor in morphology were observed as compared to the crosslinked peptide assemblies (Supplementary Information). Contrary to non-crosslinked particles, contact with the AFM-tip did not damage the crosslinked assembled structures and instead could move the spheres intact over the mica (results not shown). These findings indicate that the intermolecular disulfide crosslinks indeed preserved the peptide supramolecular structure, making them more resistant to external disruptive forces.

Atomic force microscopy of SA2C2 (A) and SA2C3 (B) crosslinked assemblies immobilized on poly(ornithine) coated mica. The scan size of the images is 1.0 × 1.0 μm and the z-scale is 16 nm.
Microscopy-Based Size Distribution
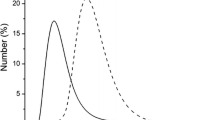
Using the data on particle size obtained with cryoTEM and AFM, a microscopy-based size distribution graph was constructed (Fig. 5). In agreement with DLS (Fig. 2), the peptide assemblies displayed a wide particle size distribution. The mean particle size as was determined by DLS, the technique that is often used to establish the particle size and size distribution (20,21), matched the largest particles present in the particle population. This is explained by the fact that this technique is exceptionally sensitive to the larger particles in a population or a cluster of particles (scattering varies with the 6th power of the radius of the particles) (Fig. 2) (22). The actual size of the particles is relevant to, for example, drug delivery applications. With respect to in vivo biodistribution kinetics, reduction of the particle size typically correlates to an enhanced penetration in diseased tissue, like tumors (23–25). Moreover, small particle sizes may facilitate the cellular entry of the drug carriers by endocytotic processes (26,27). Based on the microscopic examination it can be concluded that in number the majority (>90%) of the crosslinked oligopeptide assemblies exhibited a radius between 7 and 30 nm for the SA2C2 assemblies, and between 10 and 30 nm for SA2C3 assemblies.

Size distributions in number of the SA2C2 (A) and SA2C3 (B) crosslinked assemblies, based on cryoTEM and AFM analysis.
Calcein Encapsulation and Release
We have previously shown that non-crosslinked SA2 peptide vesicles can be loaded with calcein (10). In order to confirm that the introduction of cysteine residues in the peptide sequence does not harm self-assembly and the vesicular architecture of these oligopeptides calcein entrapment assay was performed for the SA2C2 and SA2C3 assemblies. Calcein was entrapped by hydration of the acid-precipitated peptides in a buffered calcein solution (pH 8.0). Separation of the free calcein from the calcein associated with the peptide assemblies was done on a SEC spin column (15). Eluted fractions were collected and screened for presence of calceine fluorescence. The peptide assemblies, eluting in the void volume of the column, entrapped small but measurable amounts of calcein (Fig. 6a). As a control, calcein was added to empty pre-formed peptide vesicles before separation on the column, resulting in minimal calcein co-elution (Fig. 6a). This shows that co-elution of calcein with peptide vesicles is not merely due to association of calcein with the peptide vesicles. In agreement with the observed size distribution of the assemblies (Fig. 5), the calcein entrapment data confirm the vesicular architecture. Indeed, similar surfactant-like peptides but with a different primary sequence also were demonstrated to form stable assemblies with a hydrophilic interior of radii comparable to the SA2C2 and SA2C3 assemblies (28,29).

Calcein encapsulation (A) and release (B) from the peptide assemblies. A. Crosslinked SA2C2 (triangles) and SA2C3 (blocks) assemblies prepared in the presence of calcein were separated on a size exclusion spin column. Fraction 1 and 2 represented the void volume containing the peptide assemblies. When calcein was added to preformed assembled peptide (control) no calcein elution in the void volume was observed. B. Release of calcein from the crosslinked SA2C2 (diamonds) SA2C3 (open blocks) and the non-crosslinked SA2 (filled blocks) peptide assemblies
We next tested whether inter-peptide disulfide crosslinking would change the release kinetics of calcein from these peptide vesicles. As a vesicular assembly the oligopeptides constitute a barrier towards release of small hydrophilic molecules. By covalent crosslinking this barrier function may be increased or remain unaffected, or may have decreased and become more permeable. For this, self-quenched concentrations of calcein were entrapped inside non-crosslinked and crosslinked peptide vesicles after which calcein release was monitored under sink conditions. As is shown in Fig. 6b, the release of calcein from both the SA2C2 and SA2C3 peptide vesicles showed an initial fast rate, with 50% of calcein released in 24 h, followed by a much slower release rate that continued for days. No significant differences were observed between the peptides containing 2 or 3 cysteines. Interestingly, the release of calcein from the crosslinked peptide assemblies was comparable to the non-crosslinked SA2 peptides, indicating that the covalent bonds between the peptide monomers did not increase the barrier function of oligopeptides towards small hydrophilic molecules like calcein. Evidently, the diffusion is limited if larger molecules are entrapped in the crosslinked oligopeptide vesicles and fluorescently labeled dextran 10,000, for example, diffused poorly after entrapment in the SA2C3 vesicles, over 24 h only releasing 12% of the total entrapped amount (see Supplementary information).
CONCLUSION
Although various strategies for the covalent stabilization of supramolecular assemblies have been exploited (16), disulfide bond formation as presented here is an attractive route for crosslinking self-assembled peptide vesicles. We showed that disulfide crosslinking of peptide vesicles resulted in vesicles that remained intact under conditions that disrupted non-crosslinked vesicles and liposomes. Interestingly, peptide vesicle crosslinking did not have an effect on the release kinetics of calcein, indicating that calcein release is not dependent on the exchange of peptide monomers from vesicular assemblies but driven by other as yet unclear mechanisms. The rate of calcein release from peptide vesicles as shown here is much faster compared to calcein release from liposomes. This may be explained by differences in the hydrophobic domains of liposomes compared to peptides vesicles, with the latter being able to form hydrogen-bonds with water or entrapped substrates, thereby facilitating transport of small, hydrophilic compounds over the peptide membrane. This high permeability of small substrates may be an advantage for particular drug delivery applications. For example, peptide vesicles may be ideal for delivering potentially immunogenic prodrug-converting enzymes to tumor sites without the need to artificially enhance membrane permeability as has been described for liposomes (30). In vitro evaluation of peptide vesicles as drug delivery systems is currently under way.
Abbreviations
- AFM:
-
Atomic Force Microscopy
- CAC:
-
critical aggregation concentration
- cryoTEM:
-
cryogenic transmission electron microscopy
- DLS:
-
dynamic light scattering
- DMF:
-
dimethylformamide
- DTT:
-
dithiotreitol
- PB:
-
phosphate buffer
- SUMO:
-
small-ubiquitin modifying protein
REFERENCES
Zhang S, Lockshin C, Cook R, Rich A. Unusually stable beta-sheet formation in an ionic self-complementary oligopeptide. Biopolymers. 1994;34:663–72.
Holowka EP, Sun VZ, Kamei DT, Deming TJ. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat Mater. 2007;6:52–7.
Ellis-Behnke RG, Liang YX, You SW, Tay DK, Zhang S, So KF, et al. Nano neuro knitting: peptide nanofiber scaffold for brain repair and axon regeneration with functional return of vision. Proc Natl Acad Sci U S A. 2006;103:5054–9.
Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, et al.. Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nature. 2002;417:424–8.
Rechesand M, Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–7.
Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TC, et al. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric beta-sheet tapes. Nature. 1997;386:259–62.
Aggeli A, Nyrkova IA, Bell M, Harding R, Carrick L, McLeish TC, et al. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide beta -sheet tapes, ribbons, fibrils, and fibers. Proc Natl Acad Sci U S A. 2001;98:11857–62.
Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci U S A. 1993;90:3334–8.
Zhang S, Marini DM, Hwang W, Santoso S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr Opin Chem Biol. 2002;6:865–71.
van Hell AJ, Costa CI, Flesch FM, Sutter M, Jiskoot W, Crommelin DJ, et al. Self-assembly of recombinant amphiphilic oligopeptides into vesicles. Biomacromolecules. 2007;8:2753–61.
Yangand SJ, Zhang SG. Self-assembling behavior of designer lipid-like peptides. Supramolecular Chemistry. 2006;18:389–396.
Crestfield AM, Moore S, Stein WH. The preparation and enzymatic hydrolysis of reduced and S-carboxymethylated proteins. J Biol Chem. 1963;238:622–7.
Jiskootand W, Crommelin DJ. Methods for structural analysis of protein pharmaceuticals, American Association of Pharmaceutical scientists, 2005.
Riddles PW, Blakeley RL, Zerner B. Reassessment of Ellman’s reagent. Methods Enzymol. 1983;91:49–60.
Fry DW, White JC, Goldman ID. Rapid separation of low molecular weight solutes from liposomes without dilution. Anal Biochem. 1978;90:809–15.
Hartgerink JD. Covalent capture: a natural complement to self-assembly. Curr Opin Chem Biol. 2004;8:604–9.
Butt TR, Edavettal SC, Hall JP, Mattern MR. SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif. 2005;43:1–9.
Deming TJ. Facile synthesis of block copolypeptides of defined architecture. Nature. 1997;390:386–9.
Horcas I, Fernandez R, Gomez-Rodriguez JM, Colchero J, Gomez-Herrero J, Baro AM. WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev Sci Instrum. 2007;78:013705.
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47:113–131.
Heinz L. Comparative test of methods to determine particle size and particle size distribution in the submicron range. Part Part Syst Charact. 1995;12:148–157.
Benoitand H, Froehlich D. In Huglin MB, editors. Light scattering from polymer solutions. London: Academic; 1972. pp. 520–535.
Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24:1029–46.
Heathand JR, Davis ME. Nanotechnology and cancer. Annu Rev Med. 2008;59:251–265.
Ferrari M. Nanogeometry: beyond drug delivery. Nat Nano. 2008;3:131–132.
Sheldon K, Liu D, Ferguson J, Gariepy J. Loligomers: design of de novo peptide-based intracellular vehicles. Proc Natl Acad Sci U S A. 1995;92:2056–60.
Connerand SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44.
Tsai CJ, Zheng J, Nussinov R. Designing a nanotube using naturally occurring protein building blocks. PLoS Comput Biol. 2006;2:e42.
Vauthey S, Santoso S, Gong H, Watson N, Zhang S. Molecular self-assembly of surfactant-like peptides to form nanotubes and nanovesicles. Proc Natl Acad Sci U S A. 2002;99:5355–60.
Huysmans G, Ranquin A, Wyns L, Steyaert J, Van Gelder P. Encapsulation of therapeutic nucleoside hydrolase in functionalised nanocapsules. J Control Release. 2005;102:171–9.
ACKNOWLEDGEMENTS
We thank Ronnie Willaert from the Flanders Institute for Biotechnology at the Vrije Universiteit Brussel for AFM measurements, and Fritsch Flesch for EM measurements. The Electron Microscope Facility of the Department of Biology Utrecht is acknowledged for the use of their microscopes.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Supporting information
DLS size distribution graphs, AFM micrographs and dextran 10,000 encapsulation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(DOC 108 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Hell, A.J., Crommelin, D.J.A., Hennink, W.E. et al. Stabilization of Peptide Vesicles by Introducing Inter-Peptide Disulfide Bonds. Pharm Res 26, 2186–2193 (2009). https://doi.org/10.1007/s11095-009-9933-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-009-9933-z