
Abstract
Purpose
The aim of the present study was to assess whether two widely used biomarkers for 5-HT1A-receptor mediated responses in the rat (hypothermia and corticosterone increase) could be scaled to man using allometric principles.
Materials and Methods
Mechanism-based pharmacokinetic–pharmacodynamic (PK–PD) models were developed and characterized in rats for the standard 5-HT1A-receptor agonists, buspirone and flesinoxan. Allometric scaling was investigated on the basis of simulation taking into account the inter-individual variability and clinical study design. The model-predicted effects of both flesinoxan and buspirone were compared to those published in the literature.
Results
The main finding of this analysis was that for both hypothermia and cortisol increase, the model could predict the extent of the pharmacological response in man adequately. For the hypothermic response, the time course of the response was also predicted with a high degree of accuracy. In contrast, in the case of the cortisol response, the observed time lag was, despite the fact that it fell within the model uncertainty, not predicted.
Conclusions
Based on these analyses, it is concluded that allometrically scaled mechanism based PK–PD models are promising as a means of predicting the pharmacodynamic responses in man. This approach provides for a novel way of interpreting and scaling pre-clinical pharmacological responses and ultimately facilitates the understanding and prediction of pharmacological responses in man.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Allometric scaling has been successfully applied to predict a variety of biological rates, times and dimensions (including metabolic rate, lifespan, growth rate, heart rate, DNA nucleotide substitution rates and length of aortas) between species of several orders of magnitude in body weight (for reviews see 1–3). In particular, drug metabolism is an area where methods based on allometric scaling principles have become a basis for the scaling preclinical of pharmacokinetic (PK) models to predict the time course of drug concentrations in man (4,5). It has been hypothesized (6–8) that similar methods may also be used to scale integrated pharmacokinetic–pharmacodynamic (PK–PD) models between species to predict time course of drug effects in man. However, currently there is very limited evidence that this is indeed the case. The premise for the cross-species scalability of PK–PD models is based on two fundamental assumptions, namely that (1) the model describes an essential part of the physiology of the system studied and (2) both the drug–receptor interaction and the physiological control systems are similar across the species studied. Recent work by Lepist and Jusko (7) has shown that using a relatively simple physiological model it was possible to scale both PK and PD parameters across four species. The aim of the present report was to explore the allometric scaling of the hypothermic response and the increase in corticosterone following the administration of 5-HT1A receptor agonists using two previously developed mechanism-based PK–PD models (9).
Model predictions for the hypothermic and cortisol response of the standard agents, flesinoxan and buspirone, were compared to published data. It is anticipated that the preliminary analyses presented in this study will aid the further development of allometrically scaled mechanistic PK–PD models which can be used to predict pharmacodynamic response in man. Overall, we believe that this approach provides a novel way of interpreting pre-clinical pharmacological responses which will ultimately facilitate the understanding of pharmacological responses in man.
MATERIALS AND METHODS
In vivo Pharmacological Experiments
The details of the pharmacokinetic–pharmacodynamic experiments in rats have been described previously (10,11). Briefly, 8 days prior to the experiment, the rats were operated upon. Indwelling cannulas for drug administration and blood sampling were implanted into the right jugular vein and the left femoral artery respectively. Furthermore, a telemetric transmitter (Physiotel implant TA10TA-F40 system, Data Sciences international (DSI), St. Paul MN, USA), was implanted into the abdominal cavity for the measurement of core body temperature. In the PK–PD experiments, conscious freely moving rats received an i.v. infusion of vehicle (saline) or active drug. Flesinoxan was administered in 3 mg/kg in 5 min (n = 6), 10 mg/kg in 5 and in 15 min (both n = 6) infusions. Buspirone was administered in a 5 mg/kg in 15 min (n = 6) and a 15 mg/kg in 15 min (n = 7) infusion. In each experiment in each individual rat approximately 15 to 18 serial blood samples of 50 μl were taken according to a fixed time schedule to determine the time course of the drug and corticosterone (for flesinoxan only) concentration. Body temperature was measured continuously throughout the experiment using the telemetric system. All procedures were conducted in compliance with national legislation and were ethically reviewed.
Human Data
The time courses of the pharmacodynamic responses in humans, used for comparison were taken from various publications (Table I and Fig. 1). The publications were selected because they contained a time-course of either the hypothermic or the corticosterone response to flesinoxan or buspirone. The different studies varied in design, and included open-label and cross-over studies as well as double blind and parallel run studies. The mean temperature and corticosterone versus time profiles were taken from the selected publication and were used for comparing the simulation results. Where the baseline temperature was not available a weighted average (36.51°C) was used as a substitute. Since few papers reported the actual body weight of the subjects, often healthy volunteers, a body weight of 70 kg was assumed. This is considered a reasonable assumption since the average body weight of the subjects in the studies reported by Seletti et al. (12) and Pitchot et al., (13) were 73.2 and 73.8 kg, respectively.

The temperature and cortisol responses over time for buspirone and flesinoxan in the references mentioned (Table I). Note that the lower two right panels depict cortisol data whereas the rest depicts temperature data. The numbers in the graphs represent the different doses used, where 1 represents the placebo or lowest dose used and 2 and 3 the next dose levels—please see Table I for the actual doses.
The buspirone doses in the studies available in humans were administered orally; whereas the doses tested in the rats were given intravenously. Therefore, in the simulations buspirone was administered as a 50 min infusion to represent an oral dose which reaches a Cmax at 0.8 h and the 30 mg dose was multiplied by a factor of 0.04 to reflect the absolute bioavailability of 4% (14).
Mechanism-based PK–PD Models
The pharmacokinetic and pharmacodynamic models and their analyses have been published previously (9–11). Briefly 2- and 3-compartment pharmacokinetic models were used for the pharmacokinetics of buspirone and flesinoxan, respectively. The pharmacokinetic profiles were used to quantify the relationship between the time profile of the agonist blood concentration and the time course of the hypothermic and corticosterone response. For this purpose, the previously described mechanistic PK–PD model (9) was fitted to the data on the time course of the hypothermic effect for each individual rat. In this model the hypothermic effect by the 5-HT1A-receptor agonists is considered to be the result of the attenuation of a set-point control by the drug receptor interaction. The model takes into account a zeroth order rate constant associated with the warming of the body (k in) and a first-order rate constant associated with the cooling of the body (k out). The thermostat-like regulation is implemented as a continuous process in which body temperature (T) is compared with a fixed reference or set-point temperature (T SP). 5-HT1A agonists elicit hypothermia by decreasing the baseline set-point value (T 0);
whereby the extent of the decrease is a function of the drug concentration C via the sigmoidal Emax-model.
where, E max represents the maximum stimulus the drug can produce, EC 50 is the drug concentration required to produce 50% of the maximum stimulus and n H is a slope factor, which determines the steepness of the curve. This yields the following system of equations:
in which X denotes the thermostat signal, which is driven by the difference between the body temperature T and the set-point temperature T SP on a time-scale governed by the rate constant a in Eq. 3. The thermostat signal affects the cooling of the body through an effector function \( X^{{ - \gamma }} \), which multiplies the first order rate constant k out. With four system parameters to be estimated, the degree of parameterization in Eq. 3 is high and this may lead to parameter identifiability problems. It has been shown that one parameter can be eliminated in a procedure involving the introduction of dimensionless variables (9). The procedure results in the establishment of the parameters A and B defined by,
where T 0 and X 0 are the values for T and X when no drug is present. Note that A and B represent the relative growth rate of, respectively X and T when C = 0 and T = T 0.
The plasma corticosterone concentration-time profiles were described by an indirect physiological response model (10);
where [Cort] represents the corticosterone concentration, f(C) a sigmoidal Emax-model (Eq. 1), k in is a zeroth order rate constant associated with the production of corticosterone and k out a first-order rate constant associated with the elimination of corticosterone. The pharmacokinetic and pharmacodynamic parameters are given in Table II.
Allometric Scaling of the Pharmacokinetic and Pharmacodynamic Parameters
The scaling of the physiological parameters and rate constants involves extrapolating parameters based on body weight (BW) following a simple power law, given by
where the exponent (b) generally assumes multiples of 1/4. Y is the parameter of interest and p the weight independent parameter (15). Typically b assumes the value of −1/4 for first order rate constants and the value of 3/4 for zeroth order constants (2,15). The rat pharmacokinetic parameters, clearance and volume, and the rat pharmacodynamic parameters, the first order rate constants, k out and A were scaled to obtain human parameters based on weight using the exponent, b equal to 3/4, 1 and −1/4, respectively (2,15)
The drug-related parameters potency, intrinsic activity, slope were not transformed because it is believed that affinity and efficacy are tissue and receptor dependent, both of which are expected to be similar in man and rat for the 5-HT1A receptor (89% receptor homology) (16). However, parameters like EC 50 are based on blood concentrations and may not reflect the true concentration at the site of actions. It is therefore proposed to use information from receptor binding and functional assays to supplement this information.
In a meta-analysis of various 5-HT1A receptor agonist effects on hypothermia, Zuideveld et al. (17) have shown that the affinity of flesinoxan is underestimated by approximately a factor 30. The underestimation is likely to be due to the fact that flesinoxan is subject to an active transport mechanism, resulting in a drug receptor association rate constant (Ka), estimate based on blood concentrations which may not be representative of the concentration at the site of the 5-HT1A receptor (18). The potency of flesinoxan was therefore scaled up 30 fold.
The amplification factor γ, the inter-individual variability and the residual error (proportional) were not scaled since they are dimensionless. It is acknowledged that variability may not directly translate across species. Since the PD parameters are determined in a population of Wistar rats, which may be considered less heterogeneous than the human population, this may result in an underestimation of inter-individual variability.
For the body weight of the rats a value of 0.3 kg (true body weight was 0.297 ± 0.0034 kg, mean ± SEM) was used. The scaled and non-scaled pharmacokinetic and pharmacodynamic parameters are represented in Table II.
The accuracy of the allometrically scaled pharmacokinetics was assessed by comparison with values reported in literature. Since the pharmacokinetic parameter obtained in the literature have been obtained using different models (1- versus 2- or 2- versus 3-compartmental models) and different estimation techniques (non-compartmental analysis versus non-linear mixed effects modeling) only the values of the Clearance (Cl), Volume of distribution at steady state, and terminal elimination half life can be reliably compared.
Simulations
Both mechanism-based PK–PD models were implemented in the clinical trial simulation software, Trial Simulator (v2.1.2, Pharsight Corporation, Mountain View, CA) and are available upon request. The designs of the studies mentioned was replicated as much as possible, taking into account the route of administration, the dose and the number of patients. Each study was replicated 1,000 times. The population PK–PD parameters were sampled from a multivariate normal distribution at study level, the individual parameter estimates were based on these new population parameters and a random sample from the distribution characterized by the inter-individual variability. Once sampled the parameters were scaled to a value for humans. On each measurement a proportional random error was added. Following each replicate the mean time profile, the maximal decrease (Rmax) and the slope of the decrease (Rate) for each dose-arm were calculated using S-PLUS (v6.1, Insightful Corporation, Seattle, WA). These values were then compared to observed values, allowing for assessment of the simulation performance. The 90% prediction intervals were calculated after simulation of 1,000 replicates and represent the fifth and 95th quantile.
RESULTS
Underlying the PD predictions are the allometrically scaled PK parameters. Although it is not the intent of this manuscript to address allometric scaling of PK parameters, grave errors in the PK predictions can be expected to carry through in the PD predictions. The scaling of the pharmacokinetic parameters of flesinoxan (10) and buspirone (11) resulted in estimates of the clearance of 176 and 1,050 ml/min and of the volume of distribution at steady state (Vss) of 136 and 148 L for a ‘typical’ human weighing 70 kg respectively. Bachman et al. (23) and Mahmood et al. (14) have reported a clearance of 163 and 1,980 ml/min and a Vss of 66 and 371 L for flesinoxan and buspirone respectively (see Table III for details). Although the parameters were obtained using different methods, within the context of allometric scaling the PK predictions for felsinoxan and buspirone are considered adequate.
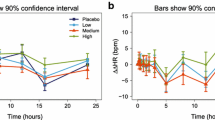
Using the allometrically scaled PK and PD parameters determined in rat, generic median predicted time course following 5 mg in a 15 min i.v. infusions of either busprione or flesinoxan for the hypothermic and the corticosterone/cortisol response respectively in man were simulated and are depicted in Fig. 2. The shaded regions denote the simulated 90% confidence interval based on the simulation of 100 subjects, thereby taking into account inter-individual variability, residual error (proportional) and parameter uncertainty. Both for the hypothermic response and for the cortisol response markedly different time-courses were predicted.

The median predicted temperature–time (a) and cortisol–time (b) profile for a human (solid white line) based on allometrically scaled parameters determined in a rat (solid black line). The shaded regions denote the simulated 90% confidence interval based on the simulation of 100 subjects (gray: humans, hashed: rats), thereby taking inter-individual variability, residual error and parameter uncertainty into account. In the example 5 mg is administered in a 15 min i.v. infusions of either busprione of flesinoxan for the hypothermic and the corticosterone/cortisol response respectively (i.e. the buspirone parameters are used for the temperature simulations and flesinoxan ones for the corticosterone/cortisol simulation). See Table II for the rats and scaled-human parameters.
Using the specific study designs, with respect to dose, number of patients, and sampling points described by the different references (Table I) the time courses of the hypothermic and, where possible, the cortisol responses were simulated. Fig. 3 depicts the simulated 90th percentile prediction intervals for the hypothermic responses in humans for buspirone and flesinoxan (top 2 rows of 4) and the simulated cortisol response for flesinoxan (hashed areas) by study and dose. The observed hypothermic and cortisol response is denoted by the filled circles and connecting line. Overall, the time course of both responses was captured well for both drugs. Only for the high dose in the cortisol response described by Seletti et al. (12) and the low dose in the study described by Pitchot et al. (13) the predictions were near the upper and lower end of the prediction interval. Following each trial replicate the maximal response (Rmax) and the Rate of reaching this maximal response was characterized in order to capture the shape of the time course numerically. This was compared to the same metric calculated from the observed data (Table IV). The maximal response was expressed as absolute value relative to the baseline (hypothermia) or as absolute value relative to placebo response (Cortisol). The predicted value represents the median prediction over 1,000 simulations and the fifth and 95th percentile represent the 90% prediction interval (90% P.I.). Overall, the time course of both responses was predicted well for both drugs, when comparing the maximal decrease (Rmax) and the Rate of decrease. Interestingly, the placebo response for the hypothermic effect in the buspirone studies was predicted poorly. Both the Rmax and the Rate were predicted to be essentially zero as no response was expected when zero drug is administered. In the studies reported by Young et al. (19) and McAllister-Williams and Massay, (20), however, a decrease in core body temperature was observed for the placebo response which explains this poor prediction. Conversely, no such placebo response was observed in the studies conducted by Seletti et al. (12) and Pitchot et al. (13). For the cortisol response the rate of the increase was respectively under and over predicted for the high and the low dose of the studies reported by Seletti et al, and Pitchot et al. Close inspection of Fig. 3 indicates a certain lag time before the cortisol response fully develops—a similar delay in the corticosterone response was not observed in rats.

Simulated 90th percentile prediction intervals for the hypothermic responses in humans for buspirone and flesinoxan (top 2 rows of 4) and the simulated cortisol response for flesinoxan (hashed areas) by study and dose (only the active doses are depicted & the title of each graph denotes refers to the applied study design). The observed hypothermic and cortisol response is denoted by the filled circles and connecting line. Note that the cortisol response has been corrected for the placebo response.
DISCUSSION
We have shown that two preclinical mechanism-based PK–PD models, one for the hypothermic and one for the corticosterone response to 5-HT1A receptor agonist, can be scaled with some degree of success to man using allometric principles (Fig. 3 and Table IV). Allometric scaling of the physiological parameters of the set-point model and the direct use of the drug related parameters as well as their variability has shown that the hypothermic response to buspirone and flesinoxan can be predicted adequately both with respect to the extent as well as the time course of the response. Allometric scaling of the corticosterone response of flesinoxan has been successful only with respect to the extent of the cortisol response. However, the time course of the cortisol response was not adequately predicted. The observed lag in the pharmacological response was not adequately explained by the indirect physiological model. The observed lag-time in humans may be explained by a control system (e.g. via hypothalamic–pituitary–adrenal axis) which is not captured by the indirect-physiological response model. It is of interest to note that this lag has not been observed in the rats. Following allometric principles, it can be speculated that the corticosterone response in rats is subject to a delay of less than 15 min. Unfortunately, the frequency of sampling of the corticosterone response in Zuideveld et al. (10) (at times 0, 10, 25, 40 min, etc.) did not allow for such a lag to be detected.
It is our view that the main reasons why the present scaling of the two mechanism-based PD models has been partly successful are that 1) the models were developed on the basis of the underlying physiology of the system under investigation and 2) that both the drug–receptor interaction and the physiological control systems are similar across the species studied. In this respect it is of interest to note that both for the hypothermic and the cortisol response, allometric scaling predicts that for humans the rate of the onset of the response is lower (reaching a maximal effect at a later time) and returns to baseline earlier (Fig. 2). In addition, the oscillatory time course of the hypothermic response in rats virtually disappears in a human which concurs with the observed clinical profiles.
The dependency of oscillatory time courses on body weight is inherent to the system. The oscillatory behavior depends on the values of the parameters in the system; scaling for larger body weights—using multiples of 1/4—results in values which do not show these typical oscillations. Allometric scaling of body temperature has often been related to allometric scaling of basal metabolic rate, which in turn has been argued to scale better with multiples of 1/3 (24). An exponent of 2/3 was first proposed by Rubner in 1883 (25) for basal metabolic rate and is in accordance with simple geometric and physiological principles if loss of temperature is assumed to be solely dependent on body surface area. In a recent paper by Chaui-Berlinck et al. (26) it is argued that whilst physical and geometrical principles are important, parameters which fall under the rein of control systems fall outside the realm of these principles. In their paper, Chaui-Berlinck et al. (26) propose a similar control system for the control of body temperature as the one in Eq. 1. Using allometric scaling Chaui-Berlinck et al. (26) hypothesize oscillations in the temperature adjustments of small homeoterms versus a graded response in large homeoterms. The current investigation therefore provides experimental evidence to this argument.
The drug related parameters E max, EC 50, n H and amplification factor γ, have not been scaled since generally serotonergic and other aminergic G-protein-coupled-receptor systems are considered to be fairly similar across both species (16). However, flesinoxan’s EC 50 required adjustment in order to adequately predict the hypothermic and cortisol response in man. In this respect it is important to note that in a meta-analysis of various 5-HT1A receptor agonist effects on hypothermia Zuideveld et al. (17) have shown that the affinity of flesinoxan is underestimated by approximately a factor 30. This apparent inconsistency can be explained by either a large difference in receptor reserve or a disproportionate difference in flesinoxan blood and effect site concentration. We believe that the latter explanation is the most likely. Specifically, a difference in receptor reserve would be expected to be consistent for both buspirone and flesinoxan. Conversely the predictions were consistently off for flesioxan’s hypothermic and cortisol response, but not for buspirone’s. Moreover, the notion that the EC 50 determined in blood might not adequately reflect the EC 50 at the effect site is supported by experiments showing that flesinoxan is subject to active transport mechanism at the level of the blood brain barrier (18). Although Van der Sandt et al. (18) have observed a mere 7–10 fold difference in total blood:brain ration between wild type and MDR 1a (−/−) mice, it is believed that differences in species, and methods used account for the differences. Therefore, based on the underestimation of flesinoxan’s affinity in vivo by a factor 30 (17), the EC 50 was adjusted accordingly. In addition to the scaling of the drug related parameters, the observed inter-individual variability on the parameters has been assumed to be similar across species. It is acknowledged that this assumption may be conservative, since the PD parameters were determined in a fairly homogeneous population of Wistar rats, with possibly a lower inter-individual variability.
In the simulations the designs of the referenced studies were imitated as much as possible, taking into account the route of administration, the dose and the number of patients. The population parameters were sampled from a multivariate normal distribution at the study level and the individual parameters were based on these new population parameters and a random sample from the distribution characterized by the inter-individual variability. Further on each measurement a random error was added in an attempt to adequately reflect the range of possible profiles. In scaling the pharmacodynamic parameters however a point estimate of the body weight was used—whereas a distribution might more adequately reflect the range of possible parameters. However since little was known about the range of bodyweights in the trials evaluated, although conservative, the use of a singe number (70 kg) was considered more appropriate.
Despite the fact that the allometric predictions rely on a number of assumptions, it is our anticipation that based on these preliminary analyses, allometrically scaled mechanistic PK–PD models could be used more widely to predict the pharmacodynamic response in man. In addition it is believed that in particular physiological systems which are conserved from an evolutionary perspective would make good candidates for allometric scaling of drug effects. The approach presented provides for a novel way of interpreting pharmacological responses in a pre-clinical setting and should ultimately facilitate the understanding of pharmacological responses in man.
References
R. H. Peters. The ecological implications of body size. Cambridge University Press, Cambridge, 1983.
K. Schmidt-Nielsen. Why is animal size so important? Cambridge University Press, Cambridge, 1995.
G. B. West and J. H. Brown. The origin of allometric scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J. Exp. Biol. 208:1575–1592 (2005).
H. Boxenbaum. Comparative pharmacokinetics of benzodiazepines in dog and man. J. Pharmacokinet. Biopharm. 10:411–426 (1982).
H. Boxenbaum. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 10:201–227 (1982).
G. A. Gronert, D. L. Fung, J. H. Jones S. L. Shafer S. V. Hildebrand, and E. A. Disbrow. Allometry of pharmacokinetics and pharmacodynamics of the muscle relaxant metocurine in mammals. Am. J. Physiol. 268:R85–91 (1995).
E. I. Lepist and W. J. Jusko. Modeling and allometric scaling of s(+)-ketoprofen pharmacokinetics and pharmacodynamics: a retrospective analysis. J. Vet. Pharmacol. Ther. 27:211–218 (2004).
K. P. Zuideveld, P. H. Van der Graaf, L. A. Peletier, and M. Danhof. Scaling pharmacodynamics from in vitro and preclinical animal studies to man: application the 5-HT1A agonists. In M. Danhof, D. R.Stanski, and P. Rolan (eds.). In 5th International Symposium on ‘Measurement and Kinetics of In vivo Drug effects’ L8:32–39 (2006).
K. P. Zuideveld, H. J. Maas, N. Treijtel, J. Hulshof, P. H. Van der Graaf, L. A. Peletier, and M. Danhof. A set-point model with oscillatory behavior predicts the time course of 8-OH-DPAT-induced hypothermia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281:R2059–R2071 (2001).
K. P. Zuideveld, A. Van Gestel, L. A. Peletier, P. H. Van der Graaf, and M. Danhof. Pharmacokinetic–pharmacodynamic modelling of the hypothermic and corticosterone effects of the 5-HT1A-receptor agonists flesinoxan. Eur. J. Pharmacol. 445:43–54 (2002).
K. P. Zuideveld, J. Rusiç-Pavletiç, H. J. Maas, L. A. Peletier, P. H. Van der Graaf, and M. Danhof. Pharmacokinetic–pharmacodynamic modeling of the hypothermic effect induced by buspirone and its metabolite 1(2-pyrimidinyl)-piperazine. J. Pharmacol. Exp. Ther. 303:1130–1137 (2002).
B. Seletti, C. Benkelfat, P. Blier, L. Annable, F. Gilbert, and C. De Montigny. Serotonin-1A receptor activation by flesinoxan in humans. Body temperature and neuroendocrine responses. Neuropsychopharmacology 13:93–104 (1995).
W. Pitchot, J. Wauthy, M. Hansenne, E. Pinto, S. Fuchs, J. Reggers, J. J. Legros, and M. Ansseau. Hormonal and temperature responses to the 5-HT1A receptor agonist flesinoxan in normal volunteers. Psychopharmacology (Berl) 164:27–32 (2002).
I. Mahmood and C. Sahajwalla. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin. Pharmacokinet. 36:277–287 (1999).
F. E. Adolph. Quantitative relations in the physiological contributions of mammals. Science 109:579–585 (1949).
P. R. Albert, Q. Y. Zhou, H. H. Van Tol, J. R. Bunzow, and O. Civelli. Cloning, functional expression, and mRNA tissue distribution of the rat 5-HT1A receptor gene. J. Biol. Chem. 265:5825–5832 (1990).
K. P. Zuideveld, P. H. Van der Graaf, D. Newgreen, R. Thurlow, N. Petty, P. Jordan, L. A. Peletier, and M. Danhof. Mechanism-based pharmacokinetic–pharmacodynamic modeling of 5-HT1A receptor agonists: estimation of in vivo affinity and intrinsic efficacy on body temperature in rats. J. Pharmacol. Exp. Ther. 308:1012–1020 (2004).
I. C. Van der Sandt, R. Smolders, L. Nabulsi, K. P. Zuideveld, A. G. de Boer, and D. D. Breimer. Active efflux of the 5-HT1A receptor agonist flesinoxan via P-glycoprotein at the blood–brain barrier. Eur. J. Pharm. Sci. 14:81–86 (2001).
A. H. Young, R. McShane, S. B. Park, and P. J. Cowen. Buspirone-induced hypothermia in normal male volunteers. Biol. Psychiatry 34:665–666 (1993).
R. H. McAllister-Williams and A. E. Massey. EEG effects of buspirone and pindolol: a method of examining 5-HT1A receptor function in humans. Psychopharmacology (Berl) 166:284–293 (2003).
I. M. Anderson and P. J. Cowen. Effect of pindolol on endocrine and temperature responses to buspirone in healthy volunteers. Psychopharmacology (Berl) 106:428–432 (1992).
T. S. Lamberg, K. T. Kivisto, and P. T. Neuvonen. Effects of verapamil and diltiazem on the pharmacokinetics and pharmacodynamics of buspirone. Clin. Pharmacol. Ther. 63:640–645 (1998).
W. J. Bachman, J. Brennan, P. Boon, A. Hage, and M. de Vries (1999) Population pharmacokinetics analysis of flesinoxan [Online]. GloboMax, 6031 University Blvd, Suite 300, Ellicott City, MD 21043 USA. http://www.globomaxservice.com/archive.htm.
C. R. White and R. S. Seymour. Mammalian basal metabolic rate is proportional to body mass2/3. Proc. Natl. Acad. Sci. U S A 100:4046–4049 (2003).
M. Rubner. Über den Einfluss der Körpergröße auf Stoff- und Kraftwechsel. Zeitschift fur Biologie 19:536–562 (1883).
J. G. Chaui-Berlinck, C. A. Navas, L. H. Monteiro, and J. E. Bicudo. Control of metabolic rate is a hidden variable in the allometric scaling of homeotherms. J. Exp. Biol. 208:1709–1716 (2005).
Acknowledgements
We would like to thank Johan Gabrielsson for the inspiring discussions on allometric scaling of pharmacodynamic responses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zuideveld, K.P., Van der Graaf, P.H., Peletier, L.A. et al. Allometric Scaling of Pharmacodynamic Responses: Application to 5-Ht1A Receptor Mediated Responses from Rat to Man. Pharm Res 24, 2031–2039 (2007). https://doi.org/10.1007/s11095-007-9336-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9336-y