
Abstract
This model proposes that the origin of life on Earth occurred as a result of a process of alteration of the chemical composition of prebiotic macromolecules. The stability of organic compounds assembled into polymers generally exceeded the stability of the same compounds as free monomers. This difference in stability stimulated accumulation of prebiotic macromolecules. The prebiotic circulation of matter included constant formation and decomposition of polymers. Spontaneous chemical reactions between macromolecules with phosphodiester backbones resulted in a non-Darwinian selection for chemical stability, while formation of strong structures provided an advantage in the struggle for stability. Intermolecular structures between nucleotide-containing polymers were further stabilized by occasional acquisition of complementary nucleotides. Less stable macromolecules provided the source of nucleotides. This process resulted first in the enrichment of nucleotide content in prebiotic polymers, and subsequently in the accumulation of complementary oligonucleotides. Finally, the role of complementary copy molecules changed from the stabilization of the original templates to the de novo production of template-like molecules. I associate this stage with the origin of life in the form of cell-free molecular colonies. Original life acquired ready-to-use substrates from constantly forming prebiotic polymers. Metabolism started to develop when life began to consume more substrates than the prebiotic cycling produced. The developing utilization of non-polymeric compounds stimulated the formation of the first membrane-enveloped cells that held small soluble molecules. Cells “digested” the nucleotide-containing prebiotic macromolecules to nucleotide monomers and switched the mode of replication to the polymerization of nucleotide triphosphates.
Similar content being viewed by others

Hypothesis
The prevailing view of the origin of life is the spontaneous coupling of prebiotic nucleobases, ribose, and (poly)phosphates into nucleotide triphosphates (NTPs), or other activated nucleoside phosphates, that subsequently polymerize into the first random ribonucleic acid (RNA) sequences. Some of these sequences might have serendipitously possessed limited self-replicating ability. Then the primitive replication began the Darwinian evolution of the sequences (Orgel 2003). This view has several conceptual problems. First, the stepwise formation of NTPs from the building blocks and their subsequent polymerization that occur during RNA biosynthesis in modern cells is unlikely to have taken place prebiotically. The appearance of the first RNA is thought to be a result of a progressive increase in complexity of the macromolecules. Secondly, selection is considered to work exclusively among replicating entities. Therefore, the onset of selection requires a high level of molecular complexity, which is unlikely to have been reached by prebiotic molecules by chance.
I propose that the origin of life lies in the non-Darwinian selection for chemical stability among prebiotic phosphodiester polymers resembling teichoic acids. The first RNA molecules might have appeared without polymerization of monomers, but rather through rearrangements of preexisting polymers. The first self-sustained system operated through self-maintenance of phosphodiester polymers, rather than through the self-replication of nucleobase sequences. The onset of selection might have preceded the onset of replication, since the longer half-life of slower decaying polymers could have provided a sufficient basis for their accumulation. Spontaneous chemical reactions between polymers with phosphodiester backbones were followed by selection for degradation-resistant products. Therefore, life could originate through the process of incomplete decomposition of prebiotic macromolecules, rather than through de novo polymerization. The accumulation of life molecules was coupled with the large-scale decay of other complex macromolecules. Thus, the overall level of order and complexity among polymers did not have to increase progressively.
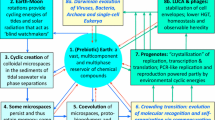
The circulation of inorganic and eventually organic compounds at the interface of atmosphere, hydrosphere, and lithosphere evidently began before the origin of life. Life arose as a part of this still ongoing geological process and substantially modified it later on. The chemical cycle of organic compounds would include the occasional formation and subsequent decay of complex polymers (Fig. 1). The spontaneous synthesis and decomposition should reach equilibrium such that a dynamic set of complex polymers of variable content persisted. Some of the polymers were formed through hydrolytically labile phosphodiester bonds between their monomeric subunits. Such macromolecules could appear in reactions between inorganic polyphosphates and compounds with multiple hydroxyl groups, such as carbohydrates or glycerol. Although similar to teichoic acids of modern Gram-positive bacteria, prebiotic random polymers should lack repeated structural units. The polyanionic phosphodiester polymers might accumulate on the surface of some minerals (Ferris 2005) that could be physically separated from energy-rich environments where polymers formed. The original phosphodiester polymers might have contained carbohydrate units of different types that were not preserved later by life.

Model for the prebiotic formation of polymers. The formation of polymers from simple molecules and polymer degradation reach equilibrium in the main cycle of the spontaneous circulation of compounds. Notably, the macromolecule formation does not represent the only sink of the available simple organic molecules. A fraction of polymers with phosphodiester backbones (PDB) escapes the main cycle. Spontaneous transesterification reactions between these polymers primarily result in the accelerated degradation of macromolecules. However, a small fraction of primary stable PDB polymers forms a separate branch of the cycle with slower decomposition kinetics. Eventually, the same mechanism leads to the accumulation of even more stable secondary PDB polymers
Certainly, macromolecules with more stable backbones would form under prebiotic conditions and become more abundant than phosphodiester polymers. However, the excessive stability would decrease the possibility of intermolecular reactions. Thus, composition of such polymers would not be substantially influenced by the presence of previously accumulated polymers. Like minerals, macromolecules with stable backbones should be an inert component of the environment. Lacking the capacity for molecular evolution, such polymers would persist without changes at the constant level over time (Fig. 1, main cycle). Glycoside and peptide bonds were likely common in prebiotic polymers. Modern enzymes catalyze transglycosylation and transpeptidation reactions, in which one glycoside or peptide bond is replaced by another (Faijes and Planas 2007; Dassa et al. 2004; Popp and Ploegh 2011). However, efficiencies of such recombination-like reactions are negligible without enzymatic catalysis.
In contrast, phosphodiester bonds in polymers allow reactions by the transesterification mechanism (Fig.2). Transesterification occurs when a hydroxyl group makes a nucleophilic attack on a phosphodiester bond to form a new phosphodiester bond while displacing a hydroxyl group (Fig. 2). In fact, the inherent chemical instability of RNA is primarily due to the spontaneous scission of phosphodiester linkages via intramolecular transesterification reactions. This scission begins with the nucleophilic attack of the neighboring phosphorus by the 2’-hydroxyl group of ribose, which cleaves the original 3’-5’ linkage and generates a new phosphodiester bond, forming the 2’-3’ cyclophosphate (Soukup and Breaker 1999). Although intermolecular spontaneous transesterification reactions are generally less efficient (Spirin 2010), an optimized in vitro transesterification protocol allows production of clearly detectable quantities of recombinant molecules (Lutay et al. 2007). Therefore, transesterification reactions between prebiotic polymers with phosphodiester bonds likely took place, thus making some polymers longer at the expense of the others.

Transesterification reaction between hypothetical irregular prebiotic precursors for self-replicating molecules. a. Ribose constitutes a small fraction of all monosaccharide units. The modules are not connected exclusively by phosphodiester bonds. Nucleobase B2 is unable to form base pairs. Nucleobases constitute a minority of attached groups, with other groups represented by R1 and R2. The primer polymer (red polygons) is bound to the template branched polymer (gray rounded rectangles) without base pairs. Base pairing B1::B3 between the template and substrate polymer aligns the primer and the substrate for the reaction (blue rectangles). Participation of the mineral surface in catalysis of transesterification is not shown. Reaction results in stabilization of the template-primer complex and the accelerated decomposition of the leftover substrate (polymer containing Sugar 7). b. Completion of the reaction (the joining of Sugar S and Sugar P) results in the recombination between the substrate and primer. The primer single-base extension would take place by subsequent hydrolysis of the phosphodiester bond between Sugar 6 and Sugar S. A cis-diol structure in Sugar S may provide an alternative chemical mechanism through a cyclic phosphate-generating cleavage of a phosphodiester bond between Sugar S and Sugar 7 followed by a ligation with the hydroxyl group from the closely spaced Sugar P (not shown, Lutay et al. 2007)
As a result, a population of more stable phosphodiester polymers could form outside of the main cycle (Fig. 1) and modify the pathways of subsequent reactions. An increasingly larger fraction of newly formed phosphodiester polymers could react with previously accumulated ones. However, the majority of products must have been less stable than the parental macromolecules, increasing the overall rate of decomposition and supplying more available subunits for spontaneous synthesis. Therefore, the composition and structure of dynamic phosphodiester polymers could be constantly influenced by the surrounding polymers with phosphodiester backbones. For any particular phosphodiester macromolecule, other phosphodiester polymers could either be protecting partners, act as substrates, or accelerate their decomposition (Fig. 2). These features opened the possibility for molecular evolution towards higher stability for a particular group of macromolecules with the intrinsically unstable backbones. Such evolution could result in quantitative growth, since the more stable molecules would accumulate at higher levels at equilibrium. Spontaneous transesterification reactions could be the chemical mechanism that modified the composition of the polymers while preserving the phosphodiester nature of their backbones. Perhaps, phosphodiester polymers were substantially less abundant than other prebiotic macromolecules. However, the absence of phosphodiester bonds in polymers with alternative backbones precluded their ability to participate directly in transesterification reactions. In polymers with mixed backbones, only the phosphodiester portion could be active (Fig. 2). In other words, phosphodiester polymers were becoming an increasingly autonomous subsystem in the environment. This primitive genetic system with a limited ability of self-reproduction would preserve the overall composition of polymers, but not their identities (New and Pohorille 2000). Evidently, life has retained the intrinsic ability of its predecessors to evolve.
Occasional modifications of some hydroxyl groups in the sugar-phosphate backbone might have screened polymers from the solvent. Modification with nucleobases would have resulted in polymers containing nucleotides. Spontaneous formation of intramolecular structures might have stabilized phosphodiester polymers by restricting their conformational variably (Soukup and Breaker 1999). Occasional base pairing between nucleotide-containing sugar-phosphates would have contributed to folding. Such random structures could not be extensive, leaving unstructured regions available for intermolecular interactions with the neighboring polymers. The increase in strength of the intermolecular complexes could stabilize both of the interacting molecules. Nucleotide-containing polymers could be stabilized by occasional incorporation of a few (or even a single) complementary nucleotide(s) to the interacting partner in the flow of stochastic transesterifications (Fig. 2). Non-nucleotide molecules lacked this ability. Other phosphodiester polymers could act as the source of the nucleotides. A sugar residue with free hydroxyl groups in the proximity of the template could serve as a primer for the incorporation. Nucleotide-specific transesterification might have been favored in particular environments composed of mineral surfaces and/or common polymers (Biondi et al. 2007).
The selectivity directed by complementary base pairing had the dual advantage of both enhanced substrate binding and the stabilization of the resulting template-product complex (Fig. 2). The external fluctuations of the environment could dissociate the template-product complex and template-directed incorporation could repeat again if the template returned to favorable conditions. Such directed cycles of nucleotide incorporation could repeat several times before the stochastic decomposition of the template or the disappearance of the transesterification-favorable environment. Such simple template-driven incorporation cannot be viewed as replication. However, this process might have been sufficient for preferential enrichment of nucleotide-containing polymers and the subsequent appearance of oligonucleotides. This mechanism could also result in the accumulation of nucleobases with the ability of base pairing. At this step, homochiral ribose-containing 5’-3’ phosphodiester polymers could accumulate if this type of backbone had an advantage over other saccharides in the template-driven acquisition of nucleotides. Alternatively, the 5’-3’ phosphodiester bonds between ribose moieties either in the product or the template favored transesterification reactions in general. The cis-diol structure of ribose might have allowed the 2’, 3’-cyclophosphate-generating cleavage of a phosphodiester bond followed by ligation with the 5’-hydroxyl group in a closely spaced ribose residue (Lutay et al. 2007). This spontaneous cleavage constitutes the chemical basis for the elucidation of RNA secondary structure by the in-line probing technique (Soukup and Breaker 1999). In each case, “the chemical properties of ribose led to its selective enrichment by some prebiotic process” (Joyce 1989). In contrast, more flexible polymers containing glycerol instead of carbohydrate modules would decompose faster.
Both the template-directed and the non-templated rearrangements participated in the formation of new polynucleotides. The template-provided “information” was limited to a single round of nucleotide incorporation. This information did not determine the entire structure of the products. The subsequent stability-driven selection among polynucleotides would have gradually resulted in the accumulation of loosely fixed sequences. Sequence information appeared in order to encode simple structural details. Polymers became less dependent on the favorable environment for templated extension. This process might have led to the appearance of the first ribozymes. Note that the majority of modern ribozymes “catalyses phosphoryl transfer reactions, either hydrolysis or transesterification” (Lilley and Sutherland 2011).
At some point, the formation of copies complementary to the regions of the template became self-sufficient for the maintenance of polymeric pools. Accumulation of complementary oligonucleotides switched the role of copy molecules from the stabilization of original templates to the de novo production of template-like molecules. Therefore, the efficiency of formation of reverse complements became more important than the stability of parental polymers. The chemistry of this process was preserved. This event can be considered as the onset of replication, Darwinian evolution, and the origin of life in the form of the “RNA world” (Gilbert 1986; Orgel 2003). The first truly replicating RNA molecules probably did not form cell-like structures, instead growing on mineral surfaces as “molecular colonies” (Spirin 2002, 2010) surrounded by prebiotic polymers. The model presented above suggests that replication was primed by other polymers. Therefore, the reverse complement copies remained attached to those primers. The limited fidelity and perhaps processivity of the original ribozymes would have allowed replication of only short fragments of the template. A fraction of the resulting fragments could associate to form functional RNA (Vaidya and Lehman 2009). Production of a large excess of waste per a small fraction of functional copies followed by the preferential recycling of the waste provided a reasonable method of original replication. Any RNA could be consumed to provide monomers for making new copies of the template in the process of the “molecular cannibalism”. In addition to the slower replication rate after mismatches (Rajamani et al. 2010), such replication would have also served to avoid error catastrophe in the highly error-prone copying of the sequence information.
Originally, the emerging RNA world did not require metabolism. Large amounts of polymers were constantly produced by the ongoing prebiotic cycling of matter. These polymers were the ready-to-use substrates for the first Darwinian molecules. Metabolism started to develop only after the advanced RNA world began to consume more substrates than the prebiotic cycling produced. The resulting competition for limited resources drove the appearance of the synthetic capacity. Each step in the first metabolic pathways arose for producing the desired compounds from the available ones after the exhaustion of the desired compounds in the environment. Perhaps, the first metabolic pathways were the conversion of nucleobases and saccharides in prebiotic phosphodiester polymers to nucleotide substrates suitable for RNA replication.
Next, the growing competition for substrates resulted in the utilization of non-polymeric compounds, including lipids. The need to hold small soluble molecules and prevent their diffusion out of RNA colonies led to the formation of the first membrane-enveloped cells (Woese 1998). Since mineral surfaces were unavailable inside cells, the catalytic activity of minerals was substituted by more complex ribozymes and cofactors. The appearance of cells allowed for the switch in the mode of RNA biosynthesis from transesterification to polymerization. Originally nucleotide monophosphate units were taken from the “substrate” phosphodiester polymers that acted as the source of both nutrients and energy. Cells “digested” the substrate, probably by pyrophosphorolysis, making the high-energy monomers (NTPs) for RNA synthesis. Later, NTPs became the major energy source for protein enzymes. The appearance of cells also stimulated amino acid metabolism that developed into protein biosynthesis and resulted in the RNA-protein world. The emergence of the first viruses followed the successful appropriation of essential resources by the cellular life that complicated RNA replication outside of cells.
The origin of life required the emergence of increasing complexity among prebiotic macromolecules. This growing complexity seemingly contradicts the second law of thermodynamics, which states that a system can spontaneously develop only to the direction of increasing entropy. My model provides an explanation for this apparent contradiction. The formation of a small pool of stable polymers by rearrangements among preexisting macromolecules was accompanied by the accelerated decomposition of a much larger pool of less stable polymers. Unstable macromolecules were actually consumed as substrates (or building blocks) for more stable polymers. The entropically unfavorable growth in the complexity was coupled to the general decrease in the level of organization. Therefore, the overall disorder in the system was increasing. My hypothesis suggests that the ancestral phosphodiester bonds provided the internal source of energy for the first replicating molecules. The model also explains why the proposed prebiotic formation of polymers no longer operates. Life developed a much more efficient ability to create and degrade polymers. Moreover, the compounds necessary for the formation of prebiotic polymers are preferentially consumed by life as nutrients.
The proposed scenario for the origin of life on Earth is consistent with computational simulations for evolution of prebiotic polymers through stability. A model was developed to examine the evolutionary potential for a system of peptides acting in a double role as catalysts and substrates of chemical reactions. This simulation revealed that the bias toward the destruction of less efficient catalysts was sufficient for the increase of the catalytic capability and complexity of the system. This increase in overall system efficiency without preservation of the identity of macromolecules was called “non-genomic evolution”. Although this model was suggested for the evolution of autocatalytic peptides, the assumptions of the model are not specific to the chemical nature of polymers as long as those polymers are capable of performing constructive and destructive processes (New and Pohorille 2000). Phosphodiester polymers are even better suited for non-genomic evolution since the energy for ligation is stored in their backbones. Another simulation indicated that UV radiation could selectively lead to prebiotic enrichment of sugar-phosphate polymers carrying nucleobases as UV protectors (Mulkidjanian et al. 2003). Note that my model suggests that the primary mechanism for the protection of phosphodiester polymers mediated by nucleobases functioned through strengthening of their structure by formation of conventional base pairs, rather than through quenching UV light.
The proposed hypothesis is subject to experimental validation. Stochastic polymers with mixed backbone might be obtained by polymerization of simple molecules, such as formaldehyde, hydrogen cyanide, and inorganic phosphate (Joyce 1989 and references therein). Intuitively, such polymers can only decay at some rate. In contrast, the model predicts a different outcome for macromolecules with phosphodiester bonds under optimized conditions. Firstly, some products of spontaneous transesterification reactions among random phosphodiester polymers (like those shown in Fig. 2) should accumulate. These products should be enriched in the ribose and nucleobase content and may be larger than the parental macromolecules. Secondly, addition of the accumulated products to fresh random phosphodiester polymers should influence the pathway of subsequent transesterifications. Some of the common earth minerals may favor these processes. Minerals can increase the local concentration of phosphodiester polymers through allowing adsorption to a solid surface (Franchi et al. 2003). Some minerals may even catalyze transesterification (Ferris 2005).
Hypothesis Summary
Formation of many different organic molecules under conditions mimicking the prebiotic Earth is well established (Miller 1952; Oro 1961), and the ability of organic molecules to form random polymers is reasonable, but not well characterized. The intrinsic tendency of large molecules to precipitate and to bind to solid surfaces decreases their exposure to the solvent. I propose that the stability of organic compounds assembled into polymers generally exceeded the stability of the same compounds as free monomers. Moreover, the extraction of organic molecules out of solvent shifted the equilibrium between formation and decomposition toward formation of additional compounds.
I hypothesize that prebiotic cycling of matter between monomers and polymers constantly produced irregular macromolecules concentrated at (and maybe catalyzed by) certain mineral surfaces. The incomplete decay of the random prebiotic polymers eventually led to the emergence of life. A fraction of polymers contained phosphodiester bonds. Spontaneous transesterification reactions that took place between such macromolecules without external energy sources shaped the content and the diversity of these polymers (Spirin 2010). These reactions represented the chemical mechanism that initiated and maintained the molecular evolution of phosphodiester macromolecules. The differences in stability among non-replicating phosphodiester polymeric molecules was the basis of the non-Darwinian selection for longer persistence. Protection of hydrolysis-susceptible phosphodiester bonds through strengthening of their structure was the direction of molecular evolution. Selected macromolecules had higher ability to destroy less stable phosphodiester polymers, but were more resistant to attacks by other molecules. Accumulation of such successive polymers accelerated decomposition of less stable prebiotic macromolecules, such as newly formed phosphodiester polymers, and fueled further selection for improved stability. This was “a period of chemical evolution, during which non-instructed processes led to progressive alteration of the chemical composition of the environment” (Joyce 1989).
Intramolecular structures limited conformational flexibility and slowed decomposition of phosphodiester macromolecules. Base-paring between the nucleotide-containing polymers further stabilized interacting partners (Soukup and Breaker 1999). This stability was also enhanced by an occasional incorporation of complementary nucleotides into a polymer templated by its interacting partner. Such incorporation resulted in the preferential enrichment of the nucleotide content in polymers and the subsequent appearance of oligonucleotides. Life began when the formation of new oligonucleotides by the template-directed transesterification mechanism became sufficient for maintenance of the oligonucleotide pool.
References
Biondi E, Branciamore S, Fusi L, Gago S, Gallori E (2007) Catalytic activity of hammerhead ribozymes in a clay mineral environment: implications for the RNA world. Gene 389:10–18
Dassa B, Yanai I, Pietrokovski S (2004) New type of polyubiquitin-like genes with intein-like autoprocessing domains. Trends Genet 20:538–542
Faijes M, Planas A (2007) In vitro synthesis of artificial polysaccharides by glycosidases and glycosynthases. Carbohydr Res 342:1581–1594
Ferris JP (2005) Genesis: rocks, minerals, and the geochemical origin of life: mineral catalysis and prebiotic synthesis: montmorillonite-catalyzed formation of RNA. Elements 1:145–149
Franchi M, Ferris JP, Gallori E (2003) Cations as mediators of the adsorption of nucleic acids on clay surfaces in prebiotic environments. Orig Life Evol Biosph 33:1–16
Gilbert W (1986) The RNA world. Nature 319:618
Joyce GF (1989) RNA evolution and the origins of life. Nature 338:217–224
Lilley DM, Sutherland J (2011) The chemical origins of life and its early evolution: an introduction. Phil Trans R Soc B 366:2853–2856
Lutay AV, Grigoriev IV, Zenkova MA, Chernolovskaya EL, Vlassov VV (2007) Nonenzymatic recombination of RNA by means of transesterification. Russ Chem Bull 56:2499–2505
Miller SL (1953) A production of amino acids under possible primitive earth conditions. Science 117:528–529
Mulkidjanian AY, Cherepanov DA, Galperin MY (2003) Survival of the fittest before the beginning of life: selection of the first oligonucleotide-like polymers by UV light. BMC Evol Biol 3:12
New MH, Pohorille A (2000) An inherited efficiencies model of non-genomic evolution. Simul Pract Theory 8:99–108
Orgel LE (2003) Some consequences of the RNA world hypothesis. Orig Life Evol Biosph 33:211–218
Oro J (1961) Mechanism of synthesis of adenine from hydrogen cyanide under possible primitive earth conditions. Nature 191:1193–1194
Popp MW, Ploegh HL (2011) Making and breaking peptide bonds: protein engineering using sortase. Angew Chem Int Ed Engl 50:5024–5032
Rajamani S, Ichida JK, Antal T, Treco DA, Leu K, Nowak MA, Szostak JW, Chen IA (2010) Effect of stalling after mismatches on the error catastrophe in nonenzymatic nucleic acid replication. J Am Chem Soc 132:5880–5885
Soukup GA, Breaker RR (1999) Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5:1308–1325
Spirin AS (2002) Omnipotent RNA. FEBS Lett 530:4–8
Spirin AS (2010) Ancient RNA World. Paleontol J 44:737–746
Vaidya N, Lehman N (2009) One RNA plays three roles to provide catalytic activity to a group I intron lacking an endogenous internal guide sequence. Nucleic Acids Res 37:3981–3989
Woese C (1998) The universal ancestor. Proc Natl Acad Sci USA 95:6854–6859
Acknowledgments
I am greatly thankful to my daughter Anastasiya Yakhnina for carefully editing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yakhnin, A.V. A Model for the Origin of Life through Rearrangements among Prebiotic Phosphodiester Polymers. Orig Life Evol Biosph 43, 39–47 (2013). https://doi.org/10.1007/s11084-012-9321-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-012-9321-2