
Abstract
The methylated derivative of l-arginine, asymmetric dimethylarginine (ADMA) is synthesized in different mammalian tissues including the brain. ADMA acts as an endogenous, nonselective, competitive inhibitor of all three isoforms of nitric oxide synthase (NOS) and may limit l-arginine supply from the plasma to the enzyme via reducing its transport by cationic amino acid transporters. Hepatic encephalopathy (HE) is a relatively frequently diagnosed complex neuropsychiatric syndrome associated with acute or chronic liver failure, characterized by symptoms linked with impaired brain function leading to neurological disabilities. The l-arginine—nitric oxide (NO) pathway is crucially involved in the pathomechanism of HE via modulating important cerebral processes that are thought to contribute to the major HE symptoms. Specifically, activation of this pathway in acute HE leads to an increase in NO production and free radical formation, thus, contributing to astrocytic swelling and cerebral edema. Moreover, the NO-cGMP pathway seems to be involved in cerebral blood flow (CBF) regulation, altered in HE. For this reason, depressed NO-cGMP signaling accompanying chronic HE and ensuing cGMP deficit contributes to the cognitive and motor failure. However, it should be remembered that ADMA, a relatively little known element limiting NO synthesis in HE, may also influence the NO-cGMP pathway regulation. In this review, we will discuss the contribution of ADMA to the regulation of the NO-cGMP pathway in the brain, correlation of ADMA level with CBF and cognitive alterations observed during HE progression in patients and/or animal models of HE.
Similar content being viewed by others

Hepatic Encephalopathy
Hepatic encephalopathy (HE) is a complex neuropsychiatric disorder that results from impaired liver function, i.e. insufficient clearance of toxins from blood, which in excess enter the brain. The impaired liver function results from acute or chronic liver failure (ALF vs. CLF) and is associated with a wide range of neurological alterations, including cognitive and motor disturbances mainly accompanying CLF [1]. A rapid progress of HE due to ALF, leads to cerebral edema and increased intracranial pressure followed by cerebral herniation and death [2].
The cellular and molecular mechanisms underlying HE are extremely complex and have not been elucidated enough, yet. However, there is a consensus that HE is mainly associated with an interference of ammonia with various aspects of brain metabolism, leading to imbalance of neural transmission [3–5]. HE is also named a primary “astrogliopathy”, because ammonia affects astrocytes, housekeepers of the central nervous system, thus impairing astrocyte-neuronal interactions, and contributing to neurotransmitter imbalance.
Dysregulation of nitric oxide (NO) production and subsequent derangement of guanidine triphosphate conversion to cyclic guanidine monophosphate (cGMP) [6, 7] is a common denominator of most of the symptoms accompanying ALF and CLF progression. At low nM concentrations, NO is an important intracellular messenger that activates soluble guanylate cyclase (sGC), initiating the cGMP production. In acute HE, ammonia-induced over-stimulation of ionotropic (mainly NMDA) glutamate receptors and activation of nitric oxide synthase (NOS) leads to an increase in NO synthesis further contributing in the generation of reactive oxygen and nitrogen species (ROS/RNS) in the brain [8–11]. On the other hand, decreased cGMP signaling in the brain has been identified as a key cause of cognitive dysfunction and memory impairment associated with chronic HE [12].
Asymmetric Dimethyl l-Arginine (ADMA), an Endogenous Nitric Oxide Synthase Inhibitor
In 1992 asymmetric (NG, NG) dimethylarginine (ADMA) was first described as an endogenous inhibitor of NOSs [13]. ADMA, its symmetric isoform (NG, NG) dimethylarginine (SDMA) and NG-monomethyl-l-arginine (monomethylarginine; l-NMMA) can regulate NO synthesis by inhibiting NOS and/or can compete for cationic amino acid transporters, which supply NOS with l-arginine [14]. ADMA is a pan-inhibitor of all three NOS isoforms, being a potent noncompetitive inhibitor of neuronal and endothelial NOS and a week inhibitor of inducible NOS. All methylated derivatives of l-arginine are ubiquitous in mammalian cells, exported from their site of origin, and imported from the plasma at distant sites by cationic amino acid transporters in exchange for l-arginine and other cationic amino acids [14, 15]. Since their discovery, the role of these compounds in the regulation of NO production has attracted increasing attention. Interestingly, next to its association with cardiovascular disease, ADMA seems also to play a role in other clinical conditions, such as critical illness, diabetes mellitus, kidney failure and hepatic failure [16, 17]. Although circulating l-arginine levels may be >100 times higher than those of ADMA, recent investigations have shown that in peripheral endothelial cells (a) intracellular ADMA: l-arginine ratio (an index of NO bioavailability) is significantly higher than the ratio measured in plasma and (b) significant NOS inhibition is achieved at physiological levels of endogenous methylarginines [18]. Faraci et al. [19] found that 50% of rat brain NOS activity was inhibited by infusion of ADMA even at low or physiological ADMA concentrations [19]. It is now well established in vitro and in vivo that micromolar concentrations of ADMA and l-NMMA, can compete with l-arginine for cell membrane transport sites. Considering that human body generates approximately 300 μmol (approximately 60 mg) of ADMA per day [14], which results in plasma ADMA concentration between 0.4 and 0.7 µM [20], and that ADMA is mainly released from myelin basic proteins highly expressed in neuronal tissue, the above evidences suggest that endogenous methylarginines may contribute to the regulation of NO levels.
ADMA Metabolism
Free methylated arginine derivatives are formed endogenously by the sequential processes of protein methylation and proteolysis by intracellular proteases and/or the proteasomal system [21]. The methylation of protein arginine residues is catalyzed by protein-methyl transferase (PRMT) family enzymes of which at least 11 mammalian isoforms have been described [22]. PRMT-1 is the main ADMA-generating enzyme. There are two known metabolic pathways for the removal of ADMA in mammals: (1) hydrolysis of ADMA to citrulline and dimethylamine in the cytoplasm by dimethylarginine dimethylaminohydrolases (DDAH-1 and DDAH-2) and (2) transamination of ADMA to α-keto-δ-(N,N-dimethylguanidino) valeric acid (DMGV) by alanine-glyoxylate aminotransferase 2 (AGXT-2) [23]. The role of the kidney and the liver in the metabolism of ADMA has been extensively studied and both organs have been proven to play a key role in the elimination of ADMA. The liver removes the majority (~80%) of ADMA exclusively via its degradation by DDAH, while the kidney uses both metabolic degradation by DDAH and urinary excretion to eliminate ADMA. DDAHs co-localize with different NOS isoforms [24], providing further indirect evidence that these enzymes may be involved in controlling the local availability of NO and downstream responses. DDAH-1 is highly expressed in the brain, suggesting its specific function in this organ. The coexistence of neuronal NOS (nNOS) and DDAH-1 in brain tissues suggests that ADMA may play some special role in the central nervous system and may be more than just an inert metabolic product. Inhibition of DDAH leads to an increase in ADMA levels and thus to a decrease in NO production. Since this pathway is regulated by complex feedback mechanisms, it probably has the ability to act as a stop signal for excessive NO production, thus potentially curbing its pathogenic action, while leaving physiological NO functions intact. Much less is known about the physiological role of AGXT-2 in ADMA metabolism. AGXT-2 is a pyridoxal phosphate-dependent aminotransferase that, in the rat, is expressed at high levels in the kidney [25] and brain [26]. AGXT-2 can also utilize ADMA as a donor of amino groups, leading to the formation of DMGV [27–29]. In this context, down-regulation of DDAH could result in an increased contribution of AGXT-2 to the metabolism of ADMA in pathophysiological conditions.
ADMA in Liver Dysfunction: Implications to the HE
A growing body of data suggests that increased concentration of ADMA, which is relatively stable and can be accurately measured in the plasma, accompanies liver dysfunctions in a wide sense and HE (for consolidated data see Table 1).
Elevated plasma concentrations of ADMA are observed in patients with severe acute alcoholic hepatitis [33] and acute liver failure [32]. In patients with compensated alcoholic or hepatitis C virus related chronic liver diseases, increased peripheral ADMA have been also reported [34, 35]. Recent data confirmed this observation in a wide cohort of cirrhotic patients [30], likewise in patients with transjugular intrahepatic portosystemic shunt (TIPS) [49].
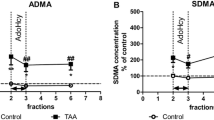
Studies on the thioacetamide (TAA)-induced rat model of ALF revealed ADMA elevation in the plasma and both in the brain cortex tissue and extracellular space with parallel lowering of liver DDAH activity [37, 38, 50]. In addition, in the BDL rat model, ADMA level significantly raises in the peripheral blood, whereas the concentration of l-arginine decreases [51]. Of note in this context, the PRMT-1 protein content was elevated in the liver of BDL rats [52, 53], but reduced in BDL rat brain [46]. There is a consensus that essential cause of ADMA elevation during liver failure is related to the lowered DDAH activity in the liver which may or may not be in line with lowered DDAH protein expression [29, 43, 54]. Recent data have revealed that DDAH-1 is predominantly present in the parenchymal liver hepatocytes while loss of protein is seen during liver fibrosis in cirrhotic patients, BDL rats and CCl4 treated rats [55].
Whether elevated ADMA concentration in the plasma can be considered a potent clinical marker of liver dysfunction and/or an accompanying factor in HE diagnosis, still remains an open question. Nevertheless, even more interesting are possible cerebral and/or systemic consequences of elevated ADMA. As already mentioned, HE is a very complex syndrome in which ADMA may exert its action in different ways, for instance by influencing vascular constriction leading to the CBF regulation, oxidative stress, cognitive function and inflammation. The authors of this review are aware that the presented list must stay “open” due to possible alternative approaches to ADMA function and ambiguously defined pathophysiological processes.
ADMA and Endothelial Function: A Contribution to the CBF Regulation
Cerebral blood flow (CBF) reflects brain energy demand and as such may be used as a potential indicator of an early decrease in brain activity. A global decrease in brain energy metabolism is one of the primary events associated with the pathogenesis of HE. Reduced cerebral oxygen consumption and CBF was observed in cirrhotic patients with an acute episode of overt HE, but not in cirrhotic patients without HE [56]. The increased CBF in cortical regions could be a common effect of the TIPS procedure, while decreased global CBF following TIPS might indicate the development of overt HE [57, 58]. Additionally, a pronounced decrease in the CBF in the cerebral cortex and whole brain was demonstrated in our laboratory in the rat TAA model of ALF [unpublished data, 59]. On the other hand, the increased CBF was reported to correlate with raised intracranial pressure and inflammatory markers in patients with ALF [60]. In general, the values of CBF reported in ALF are variable. A high CBF was demonstrated in patients with ALF in the late stage of the disease but before the development of cerebral herniation [61]. Contrary, Almdal et al. [62] reported low CBF in patients in more advanced stages of HE [62]. A study in 30 patients in various stages of HE suggested that the CBF was likely to be low [63]. Simultaneous measurement of ICP and CBF in eight patients revealed that ICP >24 mmHg was correlated with high CBF [64]. Felipo [2] in his comprehensive review presented the hypothesis that CBF was differently regulated in the cerebral cortex and cerebellum as well as at the early and late stages of HE [2]. However, this assumption is not entirely consistent with all the available data presented above.
The restriction of CBF may be one of effects of ADMA. NO is arguably the most important endogenous vasodilator regulating the perfusion of the brain, significantly influencing the tone of conductive and resistance arteries as well as venous vessels [65]. Exogenous ADMA causes concentration- and endothelium-dependent contractions of the human middle cerebral artery [66]. Similar study was conducted on rings of human middle cerebral artery from 26 autopsies, where the effects of exogenously administered ADMA were prevented by l-arginine [67]. On the one hand, ADMA might contribute to brain injury by reduction of CBF while on the other, ADMA might be involved in NOS-induced oxidative stress and excitotoxic neuronal death. After ischemic stroke, the inhibition of inducible NOS (iNOS) and nNOS have been suggested to be neuroprotective while eNOS inhibition might reduce CBF after brain injury [68]. Taken together, the effects of ADMA, which acts as a nonselective NOS inhibitor and a mediator of oxidative stress via uncoupling of iNOS and eNOS, may be multifarious, either detrimental or beneficial. The explanation of this issue requires further studies.
ADMA and Oxidative-Nitrosative Stress
A growing body of evidence suggests that methylated derivatives of l-arginine can regulate NOS-derived superoxide production by an uncoupled nNOS [69] or eNOS [70]. Oxygen species can oxidize tetrahydrobiopterin (BH4) to dihydro-(BH2), which uncouples eNOS. Since ROS may increase intracellular ADMA levels, this is a potential positive feedback mechanism to perpetuate oxidative stress [71]. However, the effects of ADMA on nNOS are different from eNOS. In the presence of BH4, superoxide production by nNOS was independently inhibited by both ADMA and l-arginine, whereas neither ADMA nor l-arginine altered superoxide formation by eNOS in the absence of BH4 [69]. It was also reported that ADMA adduction to murine epithelial cells induced rapid increases in superoxide production, inhibited NO synthesis, and caused peroxynitrite formation. These effects of ADMA were exerted via uncoupling of iNOS [72].
Considering this, it is tempting to speculate that the observed induction of oxidative stress in HE may be modulated by ADMA. In vivo evidence for ammonia-induced oxidative stress in the brain has been obtained in animal models of acute ammonia intoxication [11, 73] and in cultured astrocytes acutely exposed to ammonia in vitro [74]. Recent works also documented an induction of oxidative stress in cirrhotic rats mainly via an overproduction of superoxide associated with a significant reduction in NO bioavailability accompanying the increased levels of nitrosylated proteins [75]. Oxidative stress may directly modulate ADMA level via its impact on ADMA metabolizing enzymes. In BDL rats, elevation in plasma and hepatic ADMA levels were positively correlated with disease severity and oxidative stress markers [52]. Also, both PRMT-1 protein expression and oxidative stress markers were elevated in the liver of this model [52]. However, a study on hepatocytes did not confirm an association of PRMT-1 expression and ROS activation [54].
Previous works indicated DDAH sensitivity to oxidative stress [76, 77]. The proposed mechanism of the inhibition of DDAH activity was associated with imbalanced pro-oxidant/antioxidant state of sulfhydryl groups in the active site of the enzyme. Indeed, the expression and activity of DDAH in hepatocytes in vitro were suppressed by superoxide and H2O2 in a time-dependent manner [54]. This assumption has been confirmed by reduction of the increased ADMA level and restoration of DDAH activity after administration of compounds with antioxidant properties, such as melatonin [52], l-histidine [37] or vitamin E which suppressed hepatic ADMA level and oxidative stress determined in the hepatic circulation in the rat BDL model [53].
ADMA and Cognitive Impairment
Manifestations of intellectual dysfunction in HE patients include psychomotor slowing, impaired attention and reduced ability to perform calculations [78, 79]. As HE worsens, impairment of speech and orientation, followed by temporal and spatial disorientations appears [80]. The most comprehensive research of Bajaj et al. [30], based on various cognitive tests, reported the association of ADMA concentration with cognitive dysfunction and inflammation in cirrhosis independently of the severity of liver disease [30]. Moreover, those authors showed that ADMA levels were significantly higher in patients who developed HE after TIPS placement compared to those who remained free of HE [30]. Memory impairment was also widely described in rats with CLF [12, 81, 82]. Furthermore, there are data indicating that the glutamate-NO-cGMP pathway in the cerebellum modulates some of types of learning, particularly the ability to learn a Y maze task [2]. Therefore, the brain ADMA and its related enzymes, involved in endogenous NO production, can be a potential cause of these disturbances. Interestingly, ADMA may contribute to brain dysfunction in patients with Alzheimer’s disease and stroke [83, 84]. Elevated peripheral ADMA may play a role in spatial deficit in BDL rats. However, authors of that study also found increased plasma ADMA levels in one of the studied groups of rats without accompanying cognition impairment [23]. On the other hand, spatial memory alterations were also observed in portacaval shunt (PCS), portal hypertension and chronic TAA intoxication models in which ADMA elevation was not precisely confirmed [85].
With a high probability, cognitive deficits in HE and chronic liver disease are linked to changes in CBF [86, 87]. It is also possible that high ADMA levels are likely to uncouple eNOS, leading to superoxide generation [34, 43] and may provide an additional mechanism leading to the worsened spatial performance.
ADMA and Inflammation
Systemic inflammation is associated with enhanced plasma ADMA levels and follows endothelial dysfunction in various inflammatory diseases, such as atherosclerosis and rheumatoid arthritis (RA) [88, 89]. Higher levels of methylarginines also correlated with an increase in mortality of patients with sepsis [90]. More recently an important role of inflammation, as an accompanying factor during HE development, has been postulated [91]. Elevated blood levels of pro-inflammatory cytokines [interleukin-1b (IL-1b), interleukin-6 (IL-6), tumor-necrosis factor-alpha (TNFα)] correlate positively with the severity of HE [92–94]. ADMA levels were markedly higher in ALF patients compared to age-matched controls, and better correlated with the levels of pro-inflammatory cytokines in pre-transplantation patients undergoing hepatic venous catheterization. Following liver transplantation, both ADMA levels and pro-inflammatory markers were reduced [32]. Comparison of patients with decompensated alcoholic cirrhosis and acute hepatitis to the patients with alcoholic cirrhosis alone revealed that former ones demonstrated a much higher increase in inflammatory response markers and ADMA blood level. Furthermore, these observations were in line with down-regulation of DDAH-2 protein expression and up-regulation of PRMT-1 protein in the liver [33]. Our group showed in the TAA-induced ALF model an increase in both plasmatic/brain ADMA and TNF-α. Moreover, increase of TNF-α mRNA was observed in the brain cortex [37]. Elevated plasma and brain TNF-α level with accompanying increase of ADMA protein were also described in cirrhosis rats [43].
ADMA and Suggested Therapeutic Strategies
A few treatment strategies used to cure hypertension, chronic kidney disease, hyperlipidemia or diabetes additionally reduce the increased level of ADMA. These include inhibitors of the renin-angiotensin-aldosterone system [95, 96], statins [97], fibrates and niacin [98, 99] or thiazolidinediones [100]. Also, antioxidants [53] or aspirin [101] contribute to the regulation of abnormal ADMA level in various disorders. So far, homocysteine-lowering therapy, despite a few promising attempts, has not been very successful in reduction of ADMA [95, 102]. The linkage between anti-inflammatory drugs and ADMA lowering therapy was recently reported in RA. Three-week treatment with etanercept or adalimumab reduced in those patients ADMA level in plasma [103]. However, previous study did not reveal an impact of 18-month methotrexate or adalimumab treatment on ADMA serum levels in RA patients [104].
Supplementation of l-arginine has also been suggested to be able to eliminate the negative ADMA impact [105]. Theoretically, in the presence of pathophysiologically relevant concentrations of ADMA and physiological concentration of l-arginine, the eNOS activity decreases which results in the NO formation rates below the physiological level. In such conditions, supplementation with exogenous l-arginine displaces the competitive inhibitor and restores the physiological l-arginine/ADMA ratio [106]. l-Arginine is the principal substrate of NOS and several early studies in human and animal models reported the beneficial effects of acute and chronic l-arginine supplementation on endothelial NO production [107, 108]. However, there are inconsistent results in a clinical context. It was reported that five of 17 published human studies showed no vascular health benefits of oral l-arginine supplementation [109]. Moreover, Wilcken et al. [10] reported that l-arginine affected ADMA metabolism providing a relative stable ADMA/l-arginine ratio despite frequent changes in the plasma level of l-arginine [110]. They concluded that the regulatory role of l-arginine on ADMA might explain the unexpected results in some l-arginine supplementation studies.
Taking into consideration that intracellular ADMA is mainly regulated by PRMT and DDAH, the use of specific PRMT inhibitors or DDAH agonists might be a more reasonable therapeutic strategy. However, due to a high degree of sequence conservation across the PRMT family, creation of specific PRMT inhibitors is challenging [111]. In addition, PRMT enzymes are involved in complex cellular physiology and PRMT inhibition may give rise to side effects. The development of PRMT-1-specific inhibitors is a key objective in the search for more efficient therapeutic strategies. Initial experiments demonstrated that irreversible PRMT inhibition by S-adenosyl-l-homocysteine hydrolase blocks methylation in the cell and has both preventive and therapeutic potential in an animal model of arthritis [112]. It appears that future efficient PRMT inhibitors will rather normalize than completely inhibit the PRMT-1 function, restoring ADMA to normal levels. Since ADMA inhibits NOS activity, this could result in restoration of NO production, overcoming many important secondary effects of diseases.
The primary route of elimination of hepatic ADMA involves its hydrolysis by DDAH-1. The farnesoid X receptor (FXR) belongs to a family of nuclear hormone receptors that have an important role in maintenance of bile, lipid and glucose balance [113]. A synthetic FXR agonist was shown to significantly increase hepatic DDAH-1 gene expression in diabetic rats [114] allowing for the determination of DDAH-1 as an FXR target gene. Subsequently, further studies in rodent models of cirrhosis and hypertension have determined the efficacy of FXR agonist in increasing DDAH-1 expression [55, 115, 116]. DDAH-1 augmentation was associated with a decrease in portal pressure, reduced fibrosis and decreased hepatic ADMA levels. Furthermore, Balasubramaniyan et al. [43] demonstrated that administration of ornithine phenylacetate in the BDL model of chronic liver cirrhosis decreased the abnormal brain ADMA level by restoring DDAH-1 expression concomitantly with reduction of brain ammonia and inflammation [43].
Finally, therapeutic up-regulation of AGXT-2 may have advantages compared with the up-regulation of DDAH-1 or DDAH-2, because the latter two enzymes may exert cancer-promoting effects that are independent of ADMA [117]. Pharmacological approaches aimed to increase the activity of AGXT-2 could have potential therapeutic value in pathological conditions in which ADMA acts as a mediator of pathogenesis.
The question arises which of the above-mentioned therapeutic strategies could be beneficial in treatment of hepatic encephalopathy? Some doubts have been raised as to whether 10–24% decreases in plasma ADMA levels induced by these agents in different diseases can be beneficial. Furthermore, the increase in ADMA level in most diseases (except for renal failure and severe shock) is relatively minor and it is unclear if this is sufficient to induce a significant NOS blockade. However, any potential strategy able to lower high plasma ADMA levels should be considered beneficial in the therapy of HE patients.
Summary and Perspectives
The molecular background underlying HE is still not completely understood and current treatment is rather symptomatic than mechanism-based. The observations that elevated ADMA levels predict future outcomes in cohort studies associated with cardiovascular diseases demonstrated the potential for methylarginines to act as a marker also in liver failure accompanying HE pathology. To date only circumstantial and correlative evidences for the role of ADMA as a mediator of selected processes in HE are available (Fig. 1). The increased circulating ADMA levels may be associated primarily with endothelial dysfunction that somehow can be translated into changes in CBF considered as a causative and/or predictive factor of overt HE. However, the exact mechanism, by which direct effects of ADMA in the brain are translated into CBF changes during HE has not been elucidated in detail. Next, a direct link between increased plasma ADMA concentration and cognitive impairment cannot be definitely confirmed due to a limited number of reports and correlative assumption. Formation of NO is regulated by both l-arginine availability and the presence of the NOS inhibitor ADMA, which may be represented by their ratio (l-arginine/ADMA). However, the application of the l-arginine/ADMA ratio is much limited due to the fact that l-arginine levels vary in a wider range than ADMA levels in the circulation, and, therefore, the ratio needs not reflect the intracellular situation. ADMA appears to regulate the cellular tissue level of NO and, thus, its biological impact both by inhibiting NO production and enhancing NO bio-inactivation by ROS. The primary role of NO synthesis in the pathogenesis of HE, plus a degree of tissue/cell specificity of the enzymes controlling methylarginine levels suggest that the modulation of ADMA metabolism may be considered also as a potential target for future therapeutic interventions. However, the modulation of DDAH and/or AGXT-2 activity and/or expression is still under research. Elucidation of the significance of ADMA in HE will require a significant broadening of the scope of research.

A potential contribution of the elevated ADMA level to the cerebral impairment occurring in the HE. Acute or chronic liver failure results in the increased level of ADMA in peripheral tissues and in the brain, due to its decreased degradation by the enzyme DDAH, among other things. High level of ADMA contributes to the restriction of the cerebral blood flow, oxidative stress, cognitive impairment and inflammation
Abbreviations
- ALF:
-
Acute liver failure
- ADMA:
-
Asymmetric dimethylarginine
- BH4 :
-
Tetrahydrobiopterin
- BDE:
-
Bile duct excision
- BDL:
-
Bile duct ligation
- CAT:
-
Cationic amino acid transporter
- CBF:
-
Cerebral blood flow
- CLF:
-
Chronic liver failure
- cGMP:
-
Cyclic guanosine monophosphate
- DDAH:
-
Dimethylarginine dimethylaminohydrolase
- eNOS:
-
Endothelial NOS
- HE:
-
Hepatic encephalopathy
- iNOS:
-
Inducible NOS
- nNOS:
-
Neuronal NOS
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- PCS:
-
Portacaval shunt
- PPVL:
-
Partial portal vein ligation
- PRMT:
-
Protein arginine methyltransferase
- SDMA:
-
Symmetric dimethylarginine
- TAA:
-
Thioacetamide
- TIPS:
-
Transjugular intrahepatic portosystemic shunt
References
Prakash R, Mullen KD (2010) Mechanisms, diagnosis and management of hepatic encephalopathy. Nat Rev Gastroenterol Hepatol 7:515–525
Felipo V (2013) Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci 14:851–858
Albrecht J, Jones EA (1999) Hepatic encephalopathy: molecular mechanisms underlying the clinical syndrome. J Neurol Sci 170:138–146
Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67:259–279
Blei AT (2008) Brain edema in acute liver failure. Crit Care Clin 24(99–114):ix
Fedele E, Raiteri M (1999) In vivo studies of the cerebral glutamate receptor/NO/cGMP pathway. Prog Neurobiol 58:89–120
Madhusoodanan KS, Murad F (2007) NO-cGMP signaling and regenerative medicine involving stem cells. Neurochem Res 32:681–694
Hermenegildo C, Monfort P, Felipo V (2000) Activation of N-methyl-d-aspartate receptors in rat brain in vivo following acute ammonia intoxication: characterization by in vivo brain microdialysis. Hepatology 31:709–715
Zieminska E, Hilgier W, Waagepetersen HS, Hertz L, Sonnewald U, Schousboe A, Albrecht J (2004) Analysis of glutamine accumulation in rat brain mitochondria in the presence of a glutamine uptake inhibitor, histidine, reveals glutamine pools with a distinct access to deamidation. Neurochem Res 29:2121–2123
Kosenko E, Kaminski Y, Lopata O, Muravyov N, Felipo V (1999) Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Rad Biol Med 26:1369–1374
Kosenko E, Venediktova N, Kaminsky Y, Montoliu C, Felipo V (2003) Sources of oxygen radicals in brain in acute ammonia intoxication in vivo. Brain Res 981:193–200
Erceg S, Monfort P, Hernandez-Viadel M, Llansola M, Montoliu C, Felipo V (2005) Restoration of learning ability in hyperammonemic rats by increasing extracellular cGMP in brain. Brain Res 1036:115–121
Vallance P, Leone A, Calver A, Collier J, Moncada S (1992) Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J Cardiovasc Pharmacol 20(Suppl 12):S60–S62
Teerlink T, Luo Z, Palm F, Wilcox CS (2009) Cellular ADMA: regulation and action. Pharmacol Res 60:448–460
Leiper J, Vallance P (1999) Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res 43:542–548
Blackwell S (2010) The biochemistry, measurement and current clinical significance of asymmetric dimethylarginine. Ann Clin Biochem 47:17–28
Ferrigno A, Di Pasqua LG, Berardo C, Rizzo V, Richelmi P, Vairetti M (2015) Changes in biliary levels of arginine and its methylated derivatives after hepatic ischaemia/reperfusion. Basic Clin Pharmacol Toxicol 119(1):101–109
Schwedhelm E, Maas R, Freese R, Jung D, Lukacs Z, Jambrecina A, Spickler W, Schulze F, Boger RH (2008) Pharmacokinetic and pharmacodynamic properties of oral l-citrulline and l-arginine: impact on nitric oxide metabolism. Br J Clin Pharmacol 65:51–59
Faraci FM, Brian JE Jr, Heistad DD (1995) Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol 269:H1522–H1527
Horowitz JD, Heresztyn T (2007) An overview of plasma concentrations of asymmetric dimethylarginine (ADMA) in health and disease and in clinical studies: methodological considerations. J Chromatogr B Anal Technol Biomed Life Sci 851:42–50
McBride AE, Silver PA (2001) State of the arg: protein methylation at arginine comes of age. Cell 106:5–8
Fackelmayer FO (2005) Protein arginine methyltransferases: guardians of the Arg? Trends Biochem Sci 30:666–671
Rodionov RN, Murry DJ, Vaulman SF, Stevens JW, Lentz SR (2010) Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J Biol Chem 285:5385–5391
Tran CT, Fox MF, Vallance P, Leiper JM (2000) Chromosomal localization, gene structure, and expression pattern of DDAH1: comparison with DDAH2 and implications for evolutionary origins. Genomics 68:101–105
Tamaki T (2000) Renal microcirculation. Nihon Rinsho 58(Suppl 1):321–324
Abe M, Ochi S, Mori Y, Yamazaki K, Ishimaru T, Yoshino Y, Fukuhara R, Tanimukai S, Matsuda S, Ueno S (2014) Distribution of D-3-aminoisobutyrate-pyruvate aminotransferase in the rat brain. BMC Neurosci 15:53
Martens-Lobenhoffer J, Rodionov RN, Drust A, Bode-Boger SM (2011) Detection and quantification of alpha-keto-delta-(N(G),N(G)-dimethylguanidino)valeric acid: a metabolite of asymmetric dimethylarginine. Anal Biochem 419:234–240
Kittel A, Maas R, Konig J, Mieth M, Weiss N, Jarzebska N, Hohenstein B, Martens-Lobenhoffer J, Bode-Boger SM, Rodionov RN (2013) In vivo evidence that Agxt2 can regulate plasma levels of dimethylarginines in mice. Biochem Biophys Res Commun 430:84–89
Ogawa T, Kimoto M, Watanabe H, Sasaoka K (1987) Metabolism of NG, NG-and NG, N’G-dimethylarginine in rats. Arch Biochem Biophys 252:526–537
Bajaj JS, Ahluwalia V, Wade JB, Sanyal AJ, White MB, Noble NA, Monteith P, Fuchs M, Sterling RK, Luketic V, Bouneva I, Stravitz RT, Puri P, Kraft KA, Gilles H, Heuman DM (2013) Asymmetric dimethylarginine is strongly associated with cognitive dysfunction and brain MR spectroscopic abnormalities in cirrhosis. J Hepatol 58:38–44
Brenner T, Fleming TH, Rosenhagen C, Krauser U, Mieth M, Bruckner T, Martin E, Nawroth PP, Weigand MA, Bierhaus A, Hofer S (2012) L-Arginine and asymmetric dimethylarginine are early predictors for survival in septic patients with acute liver failure. Mediators Inflamm 2012:210454
Mookerjee RP, Dalton RN, Davies NA, Hodges SJ, Turner C, Williams R, Jalan R (2007) Inflammation is an important determinant of levels of the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine (ADMA) in acute liver failure. Liver Transplant 13:400–405
Mookerjee RP, Malaki M, Davies NA, Hodges SJ, Dalton RN, Turner C, Sen S, Williams R, Leiper J, Vallance P, Jalan R (2007) Increasing dimethylarginine levels are associated with adverse clinical outcome in severe alcoholic hepatitis. Hepatology 45:62–71
Lluch P, Torondel B, Medina P, Segarra G, Del Olmo JA, Serra MA, Rodrigo JM (2004) Plasma concentrations of nitric oxide and asymmetric dimethylarginine in human alcoholic cirrhosis. J Hepatol 41:55–59
Vizzutti F, Romanelli RG, Arena U, Rega L, Brogi M, Calabresi C, Masini E, Tarquini R, Zipoli M, Boddi V, Marra F, Laffi G, Pinzani M (2007) ADMA correlates with portal pressure in patients with compensated cirrhosis. Eur J Clin Invest 37:509–515
Nijveldt RJ, Teerlink T, Siroen MP, van der Hoven B, Prins HA, Wiezer MJ, Meijer C, van der Sijp JR, Cuesta MA, Meijer S, van Leeuwen PA (2004) Elevation of asymmetric dimethylarginine (ADMA) in patients developing hepatic failure after major hepatectomy. J Parent Enter Nutr 28:382–387
Milewski K, Hilgier W, Albrecht J, Zielinska M (2015) The dimethylarginine (ADMA)/nitric oxide pathway in the brain and periphery of rats with thioacetamide-induced acute liver failure: modulation by histidine. Neurochem Int 88:26–3138
Bekpinar S, Vardagli D, Unlucerci Y, Can A, Uysal M, Gurdol F (2015) Effect of rosiglitazone on asymmetric dimethylarginine metabolism in thioacetamide-induced acute liver injury. Pathophysiology 22:153–157
Develi-Is S, Bekpinar S, Kalaz EB, Evran B, Unlucerci Y, Gulluoglu M, Uysal M (2013) The protection by heme oxygenase-1 induction against thioacetamide-induced liver toxicity is associated with changes in arginine and asymmetric dimethylarginine. Cell Biochem Funct 31(2):122–128
Bal F, Bekpinar S, Unlucerci Y, Kusku-Kiraz Z, Onder S, Uysal M, Gurdol F (2014) Antidiabetic drug metformin is effective on the metabolism of asymmetric dimethylarginine in experimental liver injury. Diabetes Res Clin Pract 106:295–302
Ferrigno A, Palladini G, Bianchi A, Rizzo V, Di Pasqua LG, Perlini S, Richelmi P, Vairetti M (2014) Lobe-specific heterogeneity in asymmetric dimethylarginine and matrix metalloproteinase levels in a rat model of obstructive cholestasis. BioMed Res Int 2014:327537
Sharma V, Ten Have GA, Ytrebo L, Sen S, Rose CF, Dalton RN, Turner C, Revhaug A, van-Eijk HM, Deutz NE, Jalan R, Mookerjee RP, Davies NA (2012) Nitric oxide and l-arginine metabolism in a devascularized porcine model of acute liver failure. Am J Physiol Gastrointest Liver Physiol 303:G435–G441
Balasubramaniyan V, Wright G, Sharma V, Davies NA, Sharifi Y, Habtesion A, Mookerjee RP, Jalan R (2012) Ammonia reduction with ornithine phenylacetate restores brain eNOS activity via the DDAH-ADMA pathway in bile duct-ligated cirrhotic rats. Am J Physiol Gastrointest Liver Physiol 302:G145–G152
Huang LT, Hung JF, Chen CC, Hsieh CS, Yu HR, Hsu CN, Tain YL (2012) Endotoxemia exacerbates kidney injury and increases asymmetric dimethylarginine in young bile duct-ligated rats. Shock 37(4):441–448
Yang YY, Lee TY, Huang YT, Chan CC, Yeh YC, Lee FY, Lee SD, Lin HC (2012) Asymmetric dimethylarginine (ADMA) determines the improvement of hepatic endothelial dysfunction by vitamin E in cirrhotic rats. Liver Int 32(1):48–57
Huang LT, Chen CC, Sheen JM, Chen YJ, Hsieh CS, Tain YL (2010) The interaction between high ammonia diet and bile duct ligation in developing rats: assessment by spatial memory and asymmetric dimethylarginine. Int J Dev Neurosci 28:169–174
Laleman W, Omasta A, Van de Casteele M, Zeegers M, Vander Elst I, Van Landeghem L, Severi T, van Pelt J, Roskams T, Fevery J, Nevens F (2005) A role for asymmetric dimethylarginine in the pathophysiology of portal hypertension in rats with biliary cirrhosis. Hepatology 42:1382–1390
Butterworth RF, Norenberg MD, Felipo V, Ferenci P, Albrecht J, Blei AT (2009) Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int 29:783–788
Siroen MP, Wiest R, Richir MC, Teerlink T, Rauwerda JA, Drescher FT, Zorger N, van Leeuwen PA (2008) Transjugular intrahepatic portosystemic shunt-placement increases arginine/asymmetric dimethylarginine ratio in cirrhotic patients. World J Gastroenterol 14:7214–7219
Czarnecka A, Milewski K, Jaźwiec R, Zielińska M (2016) Intracerebral administration of S-adenosylhomocysteine or S-adenosylmethionine attenuates the increases in the cortical extracellular levels of dimethylarginines without affecting cGMP level in rats with acute liver failure. Neurotox Res. doi:10.1007/s12640-016-9668-7
Sheen JM, Chen YC, Tain YL, Huang LT (2014) Increased circulatory asymmetric dimethylarginine and multiple organ failure: bile duct ligation in rat as a model. Int J Mol Sci 15:3989–4006
Tain YL, Hsieh CS, Chen CC, Sheen JM, Lee CT, Huang LT (2010) Melatonin prevents increased asymmetric dimethylarginine in young rats with bile duct ligation. J Pineal Res 48:212–221
Yang YY, Lee TY, Huang YT, Chan CC, Yeh YC, Lee FY, Lee SD, Lin HC (2012) Asymmetric dimethylarginine (ADMA) determines the improvement of hepatic endothelial dysfunction by vitamin E in cirrhotic rats. Liver Int 32:48–57
Tain YL, Kao YH, Hsieh CS, Chen CC, Sheen JM, Lin IC, Huang LT (2010) Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Rad Biol Med 49:1088–1098
Mookerjee RP, Mehta G, Balasubramaniyan V, Mohamed Fel Z, Davies N, Sharma V, Iwakiri Y, Jalan R (2015) Hepatic dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis and is a target for therapy in portal hypertension. J Hepatol 62:325–331
Iversen P, Sorensen M, Bak LK, Waagepetersen HS, Vafaee MS, Borghammer P, Mouridsen K, Jensen SB, Vilstrup H, Schousboe A, Ott P, Gjedde A, Keiding S (2009) Low cerebral oxygen consumption and blood flow in patients with cirrhosis and an acute episode of hepatic encephalopathy. Gastroenterology 136:863–871
Zheng G, Zhang LJ, Wang Z, Qi RF, Shi D, Wang L, Fan X, Lu GM (2012) Changes in cerebral blood flow after transjugular intrahepatic portosystemic shunt can help predict the development of hepatic encephalopathy: an arterial spin labeling MR study. Eur J Radiol 81:3851–3856
Zheng G, Zhang LJ, Cao Y, Pan Z, Qi RF, Ni L, Shi D, Fan X, Lu GM (2013) Transjugular intrahepatic portosystemic shunt induced short- and long-term cerebral blood flow variations in cirrhotic patients: an arterial spin labeling MRI study. Metab Brain Dis 28:463–471
Pluta R, Albrecht J (1986) Changes in arterial and cerebral venous blood gases, cerebral blood flow and cerebral oxygen consumption at different stages of thioacetamide-induced hepatogenic encephalopathy in rat. Resuscitation 14:135–139
Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A (2004) Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol 41:613–620
Ede RJ, Williams RW (1986) Hepatic encephalopathy and cerebral edema. Semin Liver Dis 6:107–118
Almdal T, Schroeder T, Ranek L (1989) Cerebral blood flow and liver function in patients with encephalopathy due to acute and chronic liver diseases. Scand J Gastroenterol 24:299–303
Wendon JA, Harrison PM, Keays R, Williams R (1994) Cerebral blood flow and metabolism in fulminant liver failure. Hepatology 19:1407–1413
Aggarwal S, Kramer D, Yonas H, Obrist W, Kang Y, Martin M, Policare R (1994) Cerebral hemodynamic and metabolic changes in fulminant hepatic failure: a retrospective study. Hepatology 19:80–87
Iadecola C (1993) Regulation of the cerebral microcirculation during neural activity: is nitric oxide the missing link? Trends Neurosci 16:206–214
Segarra G, Medina P, Ballester RM, Lluch P, Aldasoro M, Vila JM, Lluch S, Pelligrino DA (1999) Effects of some guanidino compounds on human cerebral arteries. Stroke 30:2206–2210 (discussion 2210–2211)
Tain YL, Huang LT (2014) Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int J Mol Sci 15:11773–11782
Chen S, Li N, Deb-Chatterji M, Dong Q, Kielstein JT, Weissenborn K, Worthmann H (2012) Asymmetric dimethyarginine as marker and mediator in ischemic stroke. Int J Mol Sci 13:15983–16004
Cardounel AJ, Xia Y, Zweier JL (2005) Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J Biol Chem 280:7540–7549
Druhan LJ, Forbes SP, Pope AJ, Chen CA, Zweier JL, Cardounel AJ (2008) Regulation of eNOS-derived superoxide by endogenous methylarginines. BioChemistry 47:7256–7263
Sydow K, Munzel T (2003) ADMA and oxidative stress. Atheroscler Suppl 4:41–51
Wells SM, Holian A (2007) Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol 36:520–528
Sathyasaikumar KV, Swapna I, Reddy PV, Murthy Ch R, Roy KR, Dutta Gupta A, Senthilkumaran B, Reddanna P (2007) Co-administration of C-Phycocyanin ameliorates thioacetamide-induced hepatic encephalopathy in Wistar rats. J Neurol Sci 252:67–75
Jayakumar AR, Panickar KS, Murthy Ch R, Norenberg MD (2006) Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci 26:4774–4784
Gracia-Sancho J, Lavina B, Rodriguez-Vilarrupla A, Garcia-Caldero H, Fernandez M, Bosch J, Garcia-Pagan JC (2008) Increased oxidative stress in cirrhotic rat livers: a potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology 47:1248–1256
Palm F, Friederich M, Carlsson PO, Hansell P, Teerlink T, Liss P (2008) Reduced nitric oxide in diabetic kidneys due to increased hepatic arginine metabolism: implications for renomedullary oxygen availability. Am J Physiol Renal Physiol 294:F30–F37
Tain YL, Freshour G, Dikalova A, Griendling K, Baylis C (2007) Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am J Physiol Renal Physiol 292:F1404–F1410
Amodio P, Del Piccolo F, Marchetti P, Angeli P, Iemmolo R, Caregaro L, Merkel C, Gerunda G, Gatta A (1999) Clinical features and survivial of cirrhotic patients with subclinical cognitive alterations detected by the number connection test and computerized psychometric tests. Hepatology 29:1662–1667
Schomerus H, Hamster W (1998) Neuropsychological aspects of portal-systemic encephalopathy. Metab Brain Dis 13:361–377
Amodio P, Montagnese S (2015) Clinical neurophysiology of hepatic encephalopathy. J Clin Exp Hepatol 5:S60–S68
Collie A (2005) Cognition in liver disease. Liver Int 25:1–8
Monfort P, Erceg S, Piedrafita B, Llansola M, Felipo V (2007) Chronic liver failure in rats impairs glutamatergic synaptic transmission and long-term potentiation in hippocampus and learning ability. Eur J Neurosci 25:2103–2111
Miralbell J, Lopez-Cancio E, Lopez-Oloriz J, Arenillas JF, Barrios M, Soriano-Raya JJ, Galan A, Caceres C, Alzamora M, Pera G, Toran P, Davalos A, Mataro M (2013) Cognitive patterns in relation to biomarkers of cerebrovascular disease and vascular risk factors. Cerebrovasc Dis 36:98–105
Selley ML (2003) Increased concentrations of homocysteine and asymmetric dimethylarginine and decreased concentrations of nitric oxide in the plasma of patients with Alzheimer’s disease. Neurobiol Aging 24:903–907
Mendez M, Mendez-Lopez M, Lopez L, Aller MA, Arias J, Cimadevilla JM, Arias JL (2008) Spatial memory alterations in three models of hepatic encephalopathy. Behav Brain Res 188:32–40
Guevara M, Gines P, Jimenez W, Sort P, Fernandez-Esparrach G, Escorsell A, Bataller R, Bosch J, Arroyo V, Rivera F, Rodes J (1998) Increased adrenomedullin levels in cirrhosis: relationship with hemodynamic abnormalities and vasoconstrictor systems. Gastroenterology 114:336–343
Zafiris O, Kircheis G, Rood HA, Boers F, Haussinger D, Zilles K (2004) Neural mechanism underlying impaired visual judgement in the dysmetabolic brain: an fMRI study. NeuroImage 22:541–552
Antoniades C, Demosthenous M, Tousoulis D, Antonopoulos AS, Vlachopoulos C, Toutouza M, Marinou K, Bakogiannis C, Mavragani K, Lazaros G, Koumallos N, Triantafyllou C, Lymperiadis D, Koutsilieris M, Stefanadis C (2011) Role of asymmetrical dimethylarginine in inflammation-induced endothelial dysfunction in human atherosclerosis. Hypertension 58:93–98
Kwasny-Krochin B, Gluszko P, Undas A (2012) Plasma asymmetric dimethylarginine in active rheumatoid arthritis: links with oxidative stress and inflammation. Pol Arch Med Wewn 122:270–276
Mortensen KM, Itenov TS, Haase N, Muller RB, Ostrowski SR, Johansson PI, Olsen NV, Perner A, Se-Jensen P, Bestle MH (2016) High levels of methylarginines were associated with increased mortality in patients with severe sepsis. Shock 46(4):365–372
Shawcross DL, Sharifi Y, Canavan JB, Yeoman AD, Abeles RD, Taylor NJ, Auzinger G, Bernal W, Wendon JA (2011) Infection and systemic inflammation, not ammonia, are associated with Grade 3/4 hepatic encephalopathy, but not mortality in cirrhosis. J Hepatol 54:640–649
Goral V, Atayan Y, Kaplan A (2011) The relation between pathogenesis of liver cirrhosis, hepatic encephalopathy and serum cytokine levels: what is the role of tumor necrosis factor alpha? Hepatogastroenterol 58:943–948
Streetz K, Leifeld L, Grundmann D, Ramakers J, Eckert K, Spengler U, Brenner D, Manns M, Trautwein C (2000) Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology 119:446–460
Odeh M, Sabo E, Srugo I, Oliven A (2005) Relationship between tumor necrosis factor-alpha and ammonia in patients with hepatic encephalopathy due to chronic liver failure. Ann Med 37:603–612
Beltowski J, Kedra A (2006) Asymmetric dimethylarginine (ADMA) as a target for pharmacotherapy. Pharmacol Rep 58:159–178
Hov GG, Sagen E, Hatlen G, Bigonah A, Asberg A, Aasarod K (2011) Arginine/asymmetric dimethylarginine ratio and cardiovascular risk factors in patients with predialytic chronic kidney disease. Clin Biochem 44:642–646
Serban C, Sahebkar A, Ursoniu S, Mikhailidis DP, Rizzo M, Lip GY, Kees Hovingh G, Kastelein JJ, Kalinowski L, Rysz J, Banach M (2015) A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci Rep 5:9902
Yang TL, Chen MF, Xia X, Luo BL, Li YJ (2006) Effect of fenofibrate on the level of asymmetric dimethylarginine in individuals with hypertriglyceridemia. Eur J Clin Pharmacol 62:179–184
Westphal S, Borucki K, Luley C, Martens-Lobenhoffer J, Bode-Boger SM (2006) Treatment with niacin lowers ADMA. Atherosclerosis 184:448–450
Tahara N, Yamagishi S, Mizoguchi M, Tahara A, Imaizumi T (2013) Pioglitazone decreases asymmetric dimethylarginine levels in patients with impaired glucose tolerance or type 2 diabetes. Rejuvenation Res 16:344–351
Deng S, Deng PY, Jiang JL, Ye F, Yu J, Yang TL, Deng HD, Li YJ (2004) Aspirin protected against endothelial damage induced by LDL: role of endogenous NO synthase inhibitors in rats. Acta Pharmacol Sin 25:1633–1639
van Guldener C, Nanayakkara PW, Stehouwer CD (2007) Homocysteine and asymmetric dimethylarginine (ADMA): biochemically linked but differently related to vascular disease in chronic kidney disease. Clin Chem Lab Med 45:1683–1687
Spinelli FR, Di Franco M, Metere A, Conti F, Iannuccelli C, Agati L, Valesini G (2014) Decrease of asymmetric dimethyl arginine after anti-TNF therapy in patients with rheumatoid arthritis. Drug Dev Res 75(Suppl 1):S67–69
Turiel M, Tomasoni L, Sitia S, Cicala S, Gianturco L, Ricci C, Atzeni F, De Gennaro Colonna V, Longhi M, Sarzi-Puttini P (2010) Effects of long-term disease-modifying antirheumatic drugs on endothelial function in patients with early rheumatoid arthritis. Cardiovasc Ther 28:e53–e64
Bode-Boger SM, Muke J, Surdacki A, Brabant G, Boger RH, Frolich JC (2003) Oral l-arginine improves endothelial function in healthy individuals older than 70 years. Vasc Med 8:77–81
Bode-Boger SM, Scalera F, Ignarro LJ (2007) The l-arginine paradox: importance of the l-arginine/asymmetrical dimethylarginine ratio. Pharmacol Ther 114:295–306
Dubois-Rande JL, Zelinsky R, Roudot F, Chabrier PE, Castaigne A, Geschwind H, Adnot S (1992) Effects of infusion of l-arginine into the left anterior descending coronary artery on acetylcholine-induced vasoconstriction of human atheromatous coronary arteries. Am J Cardiol 70:1269–1275
Tousoulis D, Davies GJ, Tentolouris C, Crake T, Lefroy DC, Toutouzas P (1997) Effects of inhibition of nitric oxide synthesis in patients with coronary artery disease and stable angina. Eur Heart J 18:608–613
Preli RB, Klein KP, Herrington DM (2002) Vascular effects of dietary l-arginine supplementation. Atherosclerosis 162:1–15
Wilcken DE, Sim AS, Wang J, Wang XL (2007) Asymmetric dimethylarginine (ADMA) in vascular, renal and hepatic disease and the regulatory role of l-arginine on its metabolism. Mol Genet Metab 91:309–317 (discussion 308)
Bedford MT, Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33:1–13
Wolos JA, Frondorf KA, Esser RE (1993) Immunosuppression mediated by an inhibitor of S-adenosyl-l-homocysteine hydrolase. Prevention and treatment of collagen-induced arthritis. J Immunol 151:526–534
Trauner M, Baghdasaryan A, Claudel T, Fickert P, Halilbasic E, Moustafa T, Zollner G (2011) Targeting nuclear bile acid receptors for liver disease. Dig Dis 29:98–102
Hu T, Chouinard M, Cox AL, Sipes P, Marcelo M, Ficorilli J, Li S, Gao H, Ryan TP, Michael MD, Michael LF (2006) Farnesoid X receptor agonist reduces serum asymmetric dimethylarginine levels through hepatic dimethylarginine dimethylaminohydrolase-1 gene regulation. J Biol Chem 281:39831–39838
Ghebremariam YT, Yamada K, Lee JC, Johnson CL, Atzler D, Anderssohn M, Agrawal R, Higgins JP, Patterson AJ, Boger RH, Cooke JP (2013) FXR agonist INT-747 upregulates DDAH expression and enhances insulin sensitivity in high-salt fed Dahl rats. PloS One 8:e60653
Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, Elst IV, Windmolders P, Vanuytsel T, Nevens F, Laleman W (2014) Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 59:2286–2298
Hartzoulakis B, Rossiter S, Gill H, O’Hara B, Steinke E, Gane PJ, Hurtado-Guerrero R, Leiper JM, Vallance P, Rust JM, Selwood DL (2007) Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg Med Chem Lett 17:3953–3956
Acknowledgements
The study was supported by the National Science Centre Grant 2013/09/B/NZ4/00536.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Anna Czarnecka and Krzysztof Milewski have contributed equally to this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Czarnecka, A., Milewski, K. & Zielińska, M. Asymmetric Dimethylarginine and Hepatic Encephalopathy: Cause, Effect or Association?. Neurochem Res 42, 750–761 (2017). https://doi.org/10.1007/s11064-016-2111-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2111-x