
Abstract
Telomerase is perceived as an immortality enzyme that enables passing the Hayflick limit. Its main function is telomere restoration but only in a limited group of cells, including cancer cells. Since it is found in a vast majority of cancer cells, it became a natural target for cancer therapy. However, it has much more functions than just altering the metabolism of telomeres—it also reveals numerous so-called non-canonical functions. Thus, a question arises whether it is always beneficial to turn it off when planning a cancer strategy and considering potential side effects? The purpose of this review is to discuss some of the recent discoveries about telomere-independent functions of telomerase in the context of cancer therapy and potential side effects.
Similar content being viewed by others

Introduction
We still lack a specific target and capable tools to develop efficient cancer therapy. There is no universal strategy for several reasons. First, most markers are quantitative only, since cancer cells are derived from host cells (or a single host cell in fact) and are very much alike. Secondly, cancer cells are “smart” and they “learn” very fast at very different levels. They acquire an ability to drug sequestration, induce autophagy, metabolize a drug or pump it out from a cancer cell that altogether leads to a resistance to therapy.
One of the most characteristic features of cancer cells is a high proliferative potential. It is, to a certain extent, associated with telomerase expression and activity. This extraordinary enzyme is able to overcome limitations in cell divisions that lead to cells immortality [1]. Thus, it appears to be a valuable target in cancer therapy. However, there are a couple of reasons why caution should be exercised when aiming at telomerase. It is expressed (and/or active) not only in cancer cells. Thus, blocking the activity or expression of this immortality enzyme might provoke some rather unexpected side effects. Especially since telomere rebuilding and providing cell immortality is not the only role of this enzyme.
Telomerase activity/expression is probably only a secondary to carcinogenesis. Thus, it seems that its restoration in other, i.e., normal cells, should be safe and beneficial when giving normal cells an ability to divide and, consequently, live forever (or just significantly longer). Altogether it looks that telomerase is a relatively specific target that could be (and in fact it is) used in cancer therapy but could also be used for lifespan extension. However, it is not only about making life longer but also of better quality. There are still things we do not know about cancer and telomerase, and we also need to face the truth that longer life means longer exposure to harmful factors, accumulation of errors, mutations, etc. Thus again, it brings us to a simple and trivial message that prevention and avoiding disease is much better than facing a therapy challenge. For many reasons prevention is not always workable of course which makes a personalized diagnostics and medicine more desired. Due to the complexity of cancer cell metabolism, it seems that the only option is to study the enemy and learn not only how to defeat it but maybe also how to mimic pro-survival mechanisms.
Telomerase and telomeres
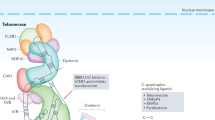
Telomerase complex
The crucial units that function as the core telomerase complex are human telomerase reverse transcriptase (hTERT) and human telomerase RNA component (hTERC). They are minimal and sufficient subunits to sustain telomerase activity in vitro [2]. However, telomerase is much more complex concerning the way it is built and, consequently, the way it is regulated. Whole, in vivo, telomerase complex is a ribonucleoprotein consisting of RNA particle (hTERC) and protein element (hTERT) as well as additional proteins including NOP10, NHP2, GAR, and dyskerin, which bind to hTERC to stabilize the complex. Telomerase builds telomeric DNA with tandem hexameric repeats (TTAGGG) that is capped by six shelterin proteins, i.e.: TRF1, TRF2, RAP1, TPP1, POT1, and TIN2 [3]. Consequently, more factors mean more complicated relations and regulations at different levels.
Telomerase is known for its ability to provide eukaryotic cells immortal via restoring chromosome ends that is considered a canonical function of the enzyme. Telomeres are perceived as a molecular clock that determines the longevity of cells allowing them to divide up to approximately 50 times (Hayflick limit) and that number is limited due to a replication-end problem [3]. Some types of cells give up to this rule (most normal cells), but some cells (a vast majority of cancers) unfortunately learned to deal with this issue via telomere restoration with telomerase (or another, rare but decently efficient, alternative lengthening of telomeres/ALT system) [4].
However, longevity is not always a good perspective. It may result in an accumulation of mutations and alterations in cells (which takes place also in cancer cells in fact) that constantly divide instead of being terminated. In most somatic cells, when a critical length of telomeres is achieved, the host cell is shifted to replicative senescence and eventually to death [5]. But before that, telomeres prevent chromosomes from joining or being recognized as a DNA double-strand break which would alert and start DNA repair mechanisms. Alternatively, dysfunctional telomeres may also cause inappropriate fusions and consequently dicentric chromosomes [5].
Telomerase expression (individual subunits) and activity are limited (with time and place) to a certain kind of cells including stem cells, activated lymphocytes, embryo, cancer, and cancer stem cells. It is mainly based on the tissue-specific activation of promoters. Additionally, telomerase plays other than canonical (telomere-related) i.e. non-canonical (telomere-independent) functions. That triggers a question if an anticancer approach should be based on turning off the enzyme since we cannot fully anticipate the consequences?
Interestingly, there are reports indicating that telomere dysfunction contributes to the pathogenesis of a variety of human cancers. Noteworthy, we do not understand this mechanism yet, and it is still disputable whether telomerase reactivation is a cause or a consequence of carcinogenesis [6,7,8,9,10]. It was also suggested that hTERT polymorphisms (e.g., rs2736098) [11] or mutations [12] might correlate with an increased cancer risk that might be a consequence of genome/chromosome increased instability. That brings us to maybe not a very straight-forward but plausible conclusion, that telomerase inhibition/repression may not always be beneficial. At least in normal cells. Thus, recognizing a complex association between telomeres, telomerase and cell metabolism we should carefully determine ways of their modulation.
What is canonical and what is not
The main canonical function of telomerase concerns the mechanism of telomere maintenance. Thus, the enzyme (if present) protects telomeres from shortening during cell division (eliminates the end-replication problem) and chromosomes from a junction. Consequently, telomerase plays a crucial role in cancer metabolism, senescence, and degenerative diseases. But there is much more than that. It appears that telomerase function cannot be attributed to nuclear localization only. And here starts the list of other, telomere-unrelated functions of hTERT and telomerase (Table 1). And this so-called non-canonical activity of telomerase contributes to the regulation of key metabolic mechanisms in cells including cell survival, gene expression, signal transduction pathways, mitochondrial metabolism and, consequently, stress response. As reported, in some cases telomerase catalytic activity is not even a prerequisite for all those effects [13].
Importantly, hTERT is also found not only in nuclear localizations but also in cytoplasm and mitochondria. Interestingly, the mitochondrial location was demonstrated to correlate with a diminished ROS levels and enhanced respiration. Moreover, hTERT was also shown to be able to bind mitochondrial DNA and tRNA. Additionally Haendeler et al., demonstrated a positive correlation of the key telomerase subunit with increased mitochondrial membrane potential [16]. Altogether, those observations led to the conclusion that some telomere-unrelated activity of telomerase might affect the resistance of cancer cells to drugs (possibly by affecting DNA damage/repair system) [16]. Consequently, understanding those complex associations may be beneficial in cancer strategy development (Tables 1, 2).
What do we learn from mouse model studies
Telomerase and telomere function is studied in some model animals including zebrafish [17], chicken [18], pig [19] or dog [20]. Noteworthy, telomeres were discovered to provide chromosomal integrity and genome stability in maize [21]. But in fact, studies using mouse model are the most promising concerning how similar these animals are to the human in the context of telomere metabolism [22, 23]. It was demonstrated in numerous studies that cellular responses to telomere dysfunction are fundamentally conserved in both species. It is also suggested that close correlation between telomerase regulation and telomere length in somatic cells might be crucial for maintaining a balance between normal metabolism, aging, and cancer. Noteworthy, telomeres found in laboratory mice are significantly longer than in humans (25–40 vs. 10–15 kb, respectively). It does not obviously change anything about the difference in our lifespan (~ 30 times shorter than in humans). Anyway, the telomerase-deficient mouse model is extremely useful in providing detailed research into the consequences of telomere dysfunction including aging, genome destabilization and cancer in mammals. Especially since in both species telomere dysfunction causes similar molecular effects including p53 pathway involvement [22].
With aging, telomeres shorten. The rate of this loss, concluding from in vitro studies of human fibroblasts or lymphocytes is very similar to a mouse model and is approximately 50–100 bp per cell division in both species [24, 25] while the telomeric sequence is also the same, i.e., TTAGGG (n). The main difference that was observed between those two species is the fact that telomerase activity and gene expression of mouse Tert (mTert, mouse telomerase reverse transcriptase) appear to be less stringently regulated in murine somatic cells [22]. Anyway, in both cases, hTERT and mTert expression are the critical determinants of telomerase activity since the expression of these subunits correlates positively with the distribution of telomerase activity in various cells [22, 26]. As shown in a mouse model that lacks the critical mRNA of the mTerc subunit, the animals are viable, fertile and had no significant morphological abnormalities [22]. As reported, it took six generations to achieve sufficient telomere attrition model displaying a subset of aging phenotypes, including alopecia, hair graying, reduced stress response and decreased longevity [27]. Xie et al., demonstrated that telomerase inhibition might contribute to telomere length-independent acceleration of aging and aging-related diseases [28]. Those observations were in concordance with previous studies revealing telomeres and telomerase role in genomic instability followed by chromosomal fusions and increased cancer incidence [1]. Additionally, the contribution of telomerase to DNA damage repair mechanism (ATM and ATR-related) as well as PARP pathways was also revealed in human cells [29, 30].
Telomerase in tumor initiation and progression
There is a debate on telomerase role in carcinogenesis and cancer progression. This enzyme is especially important to target in cancer therapy since its activity and/or expression is predominantly found in cancer cells (over 90% of cancers). However, since telomerase contributes to genome stabilization (or at least its deficiency provokes genome destabilization), it is still a challenging aspect not to provoke more genome destabilization (resulting from telomerase downregulation in non-target tissues) that might lead to cancer development [1] that was actually suggested when human p53 deficient/mutant cells were assessed [31]. However, the mechanisms governing hTERT expression in cancer remain unclear. Most reports indicate that telomerase restoration is just a secondary process accompanying carcinogenesis. However, most transcription factors that control hTERT promoter are widely expressed that makes the explanation of this association more difficult [32].
Recent observations suggest that one of the key factors in telomerase recurrence is associated with mutations in hTERT promoter upstream from the ATG start site (C228T and C250T). These mutations occur in a vast majority of cancers including melanomas (~ 70%), glioblastomas (80–90%), hepatocellular carcinomas (~ 60%), bladder cancers (~ 60%), basal cell carcinomas (~ 70%), oligodendrogliomas (~ 70%) as well as cutaneous squamous cell carcinomas (~ 50%) and thyroid cancers (up to 30%) [33]. Since they were detected across all stages and grades of cancer, it is suggested that mutations in hTERT appear as early events in carcinogenesis. Importantly, some other, but less influential, mutations were identified too. Other authors show that epidermal growth factor (EGF) may also significantly contribute to hTERT promoter activation [34]. Another factor that might control hTERT is alternative splicing of hTERT mRNA [35], but this association remains elusive. Altogether, it is still not clear how hTERT activation contributes to carcinogenesis initiation and what other mechanisms (genetic and epigenetic) are involved in this process. Similarly, we still do not know whether telomerase expression has any oncogenic potential or is simply mandatory for tumor growth support. There is no doubt however that a protein that is a promising cancer therapy target may significantly contribute to the protection of normal cells degradation/degeneration.
ALT versus telomerase
Most human cancers show telomerase expression (80–90%). Those who do not, may use alternative lengthening of telomeres (ALT) to maintain telomeres above a critical length, but there are also some cell types that utilize both mechanisms [36]. Telomerase restoration is a critical occurrence that supports carcinogenesis via overcoming the end replication problem. Consequently, it makes tumor progression possible [37]. There is some specificity concerning the way both mechanisms function. ALT-positive tumors usually are derived from mesenchymal tissues while telomerase-positive tumors are mainly derived from epithelial tissues. Importantly, telomere length in those cells is very heterogeneous. Another characteristic feature of those cells is the presence of Promyelocytic Leukemia (PML) bodies [38]. However, recent data implicate that the ALT might be less efficient than telomerase in providing tumor progression (especially metastasis). Those studies suggest that some non-canonical action of hTERT is required even if ALT works for telomere maintenance [39, 40]. It was confirmed by the observation that in ALT cells some dysfunctional telomeres exist [41]. Thus, if ALT is not sufficient for telomere maintenance, it is suggested that inhibition of telomerase might shift the majority of initially telomerase positive tumors to ALT-positive and eventually eliminate them. Consequently, repressing hTERT may be beneficial for two reasons – it would eliminate telomerase- and ALT-positive cells. It is of high importance especially, since repression of both, hTERT or ALT, results in similar effects, i.e., senescence and/or cell death [38, 42].
Functional aspects
Telomerase and proliferation
Telomerase is associated with proliferation potential that might be perceived as a positive aspect in the context of regeneration. But it is not always a beneficial feature. As demonstrated by Oh et al., overexpression of mTert in the heart (mouse model) resulted in hyperplasia and hypertrophy of cardiac myocytes. Expression of mTert also contributed to apoptosis suppression in those cells which might indicate the mTert association with proliferation. Noteworthy, this effect was observed only when a functional mTert sequence was overexpressed [43].
Additionally, it was demonstrated that the median lifespan of mTert-transgenic mice was significantly increased (up to 24% when applied in adult, 1-year old animals). Additionally, studied mice demonstrated significantly improved markers of health, fitness, and aging. Noteworthy, individuals overexpressing mTert did not develop more neoplasms comparing to their control littermates. Thus, an anti-aging property of telomerase knock-in in adult/old mammals was suggested [23].
However, there also some alternative reports showing that telomerase overexpression in basal keratinocytes leads to an increased incidence of carcinogen-induced skin tumors in mice [39]. Other studies revealed that telomerase overexpression in mammary glands of mice promoted the development of spontaneous cancers [44]. There are also some alarming reports concerning human cells showing that ectopic expression of hTERT in mammary cells (with downregulated p16INK4A) may contribute to increased resistance to growth inhibition by transforming growth factor-beta (TGF-β) [45]. It was supported by a report showing that ectopic expression of functional hTERT promoted cellular growth in primary human mammary epithelial cells (HMEC) in a mitogen-depleted environment [46]. Other authors showed that hTERT constitutive expression in HMEC led to the induction of several growth-promoting genes, including the epidermal growth factor receptor (EGFR), and downregulation of proapoptotic genes [47]. Thus, another function of telomerase (in a telomere-independent way) in regulating cellular proliferation by modulating growth-promoting genes, was shown. Interestingly, the contribution of telomerase to proliferation engages also Wnt signaling pathway that will be described further.
Telomerase translocation into mitochondria
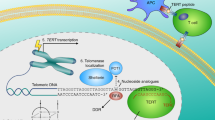
It is known that telomere shortening may directly lead to aging, but this process may also be initiated by other factors including oxidative stress, inflammation, and infections [48]. This brings a conclusion that those processes might be associated. However, we still do not know why is telomerase function so important in attenuating oxidative stress effects. It may result from the fact that telomeres are the preferred targets of oxidative insult. And this, consequently, may be related to their DNA composition, rich in guanine residues [49]. It is known that the high content of guanine is associated with generation of 8-oxoguanine (8-oxoG) species which, if not repaired, lead to DNA strand breaks, mutations or genomic instability [50]. Thus, a question arises whether it is possible that hTERT constitutes a “rescue system”, also in mitochondria? (Fig. 1).

hTERT rescue system pathways. Telomerase catalytic subunit responds to extra and intracellular signals. It may affect certain genes expression as well as protect mitochondrial metabolism. Altogether, it shows pro-survival properties
Noteworthy, some fraction of hTERT (10–20%) is localized in mitochondria probably as a result of oxidative stress [51] which suggests a protective role of the catalytic telomerase subunit. It was estimated that under normal conditions, mitochondrial hTERT localization limits the UV and ethidium bromide (EtBr) induced mtDNA damage. Additionally, the catalytic activity of the enzyme is required to exert this protective activity and hTERT was shown to bind the mtDNA in the regions encoding the ND1 and ND2 that mediate ROS generation [16].
It was also shown that cancer cells with dysfunctional telomerase were more sensitive when exposed to irradiation (carbon-ion). Detailed studies revealed that hTERT suppression caused (irrelevant to the way of inhibition/downregulation) impaired mitochondrial function. This dysfunction was manifested by decreased membrane potential and alterations in mtDNA copy number, mitochondrial mass, total ATP levels and elevated reactive oxygen species (ROS) [52]. It was also reported that mitochondrial hTERT protected these organelles from H2O2 influence and irradiation-induced DNA damage foci in the nucleus [53]. Similarly, Muzza et al. reported that the mitochondrial hTERT localization was significantly higher in tumors when exposed to high oxidative stress (H2O2) while its nuclear expression was similar in tumor and normal tissues [54]. Interestingly, expression of a mutated hTERT (unable to be translocated to mitochondria) was associated with increased levels of ROS accompanied by mitochondrial dysfunctions [55].
Mitochondrial translocation of hTERT seems to be particularly important in multidrug-resistant (MDR) cells since those two phenomena may be associated [56, 57]. Another study revealed that hTERT over-expression in mitochondria in hepatocellular carcinomas significantly increased resistance to cancer drugs, both in vitro and in vivo. A higher mitochondrial membrane potential inhibited apoptotic pathway, and an increased mtDNA copy number were observed after drug administration [58]. In another study, a neuroprotective role of hTERT in a telomerase activity-independent way was shown due to its increased mitochondrial localization [59]. Generally, a neuroprotective role of hTERT is postulated due to the induction of cellular proliferation, anti-apoptotic effects, mitochondrial stabilization, and anti-aging and anti-oxidant effects [60]. And finally, it was shown that hTERT could protect neonatal mitochondrial DNA (mtDNA) from oxidative damage during pregnancies [61]. However, there is still no confirmation whether hTERT is obligatory for normal function of mitochondria or is it recruited in an emergency/stress situation.
hTERT in neurons/nervous system
Telomerase catalytic subunit is also studied in the context of the neuroprotective activity. González-Giraldo et al., reported significant differences in hTERT expression between controls (higher) and patients (lower) with the major depressive disorder [62]. In the same paper, authors postulated association of hTERT with ROS reduction and DNA protection in mitochondrial neurons [62]. Similarly, it was demonstrated that hTERT reduction caused an increased ROS generation and oxidative damage induction by pathological tau protein (microtubule-associated protein, crucial in the pathogenesis of many human neurodegenerative diseases) in neurons [63, 64]. Thus, discovering new telomerase activating compounds seems to be a promising strategy in preventative treatments for neurodegenerative diseases, reducing disease symptoms or even extending lifespan as demonstrated in a mouse model of amyotrophic lateral sclerosis (ALS) [63, 65].
Neuropsychiatric disorders including depression, schizophrenia, anxiety, and affective disorder are also associated with telomerase shortening. There is also a hypothesis that oxidative stress and inflammation affect the structure of the replication fork in the area of telomeres [66].
Studies performed in Purkinje neurons revealed that hTERT expression was significantly increased in response to stress (i.e., X-ray radiation and a high glutamate concentration). Interestingly, the induced telomerase expression was observed in the nucleus of cells exposed to radiation while glutamate treatment induced hTERT levels in mitochondria [67]. Nevertheless, telomerase seems to play a pivotal role in the adaptive response of neurons to different types of stress.
Remote control effect
One of the most surprising facts concerning telomerase was revealed by Gutkin et al., who demonstrated that hTERT mRNA was capable of shuttling from cancer cells into telomerase negative fibroblasts via exosomes. As it appeared, in the new target cells, hTERT was translated into a fully active enzyme creating nonmalignant, telomerase positive cells [68]. Consequently, recipient fibroblasts demonstrated increased proliferation properties, extended lifespan, and senescence postponement. Additionally, target cells became resistant to DNA damage induced by phleomycin and to apoptosis [68]. Altogether it looks that telomerase translocation has a potential to affect local microenvironment.
hTERT and vascular system
hTERT was also reported to induce vascular epithelial growth factor (VEGF) expression via interaction with the gene’s promoter and transcription factor Sp1—consequently, it may stimulate angiogenesis. Studies performed in a mouse model revealed that deletion of the mTert gene leads to a limited tumor growth accompanied by reduced VEGF expression in the first generation of mTert null mice. Interestingly, studies performed in human gastric tumor samples revealed a correlation of hTERT expression levels with VEGF [69]. Similarly, telomerase inhibition in vascular endothelial cells was shown to disrupt angiogenesis and tumor growth in glioblastoma xenografts [70]. George et al., showed that hTERT downregulation (RNAi) and IFN-gamma treatment in human glioblastoma cell lines (SNB-19 and LN-18) significantly inhibited angiogenesis and tumor progression [71].
Interestingly, Falchetti et al., revealed that telomerase inhibition in Human umbilical vein endothelial cells (HUVECs) resulted in a reduction of tubule formation and cell survival in the tumor xenografts. This observation suggested that glioblastoma multiform tumor angiogenesis may be associated with telomerase upregulation [70]. This hypothesis was confirmed in HeLa cells since hTERT was shown capable of VEGF transcription induction [72].
hTERT, adhesion, and migration
As demonstrated, telomerase contributes or is associated with many different mechanisms that accompany telomerase overexpression or downregulation. Among well-known telomerase modulators, TMPyP4 is found (telomerase inhibitor). Additionally, this compound alters telomerase expression. Consequently, it provokes some difficulties in understanding the function of telomerase activity and/or expression and its association with molecular pathways. However, as demonstrated, it may also alter cell adhesion. Scratch-wound and transwell assay indicated that the compound at low levels (≤ 0.5 μM) increased cell-matrix adhesion and promoted the migration of tumor cells. In contrast, higher concentrations of TMPyP4 (≥ 2 μM) were demonstrated to inhibit cell proliferation and induce cell death [73]. This implicates an important role of telomerase modulators (both, activity and expression) in cancer cell adhesion, migration and, consequently, invasiveness potential. Similarly, adhesion experiments revealed that induced hTERT expression significantly increased cell adhesion supporting a non-canonical function for hTERT in promoting tumorigenesis [74]. Interestingly, it was also shown that hTERT affected the expression of numerous genes in different cell lines, implying that the process is indirect and cell type dependent. Importantly, the functional analysis revealed cell adhesion-related genes that were affected by hTERT in studied cell lines [74].
Gene expression
hTERT in gene expression regulation
Nmerous reports indicate that the hTERT subunit is capable of performing even much wider spectrum of activity including remodeling chromatin structure, and contribution to signaling pathways, e.g., the Wnt/β-catenin or NFkB. Those key signaling pathways constitute control cell survival and proliferation, cell polarity, differentiation during embryonic development, carcinogenesis but also migration and regeneration [75, 76]. In genome-wide transcriptional studies, it was demonstrated that hTERT enhanced epithelial cell proliferation by regulating gene expression in a similar way to Myc and Wnt controlled pathways [44]. Park et al. revealed that hTERT acted as a cofactor in the β-catenin transcription complex. It was shown that hTERT interacted with BRG1, a chromatin remodeling factor, to regulate the Wnt/β-catenin signaling pathway [77]. However, studies made by Blackburn et al. (performed in MCF7 and HeLa cells) and by Liu et al. revealed no physical association of hTERT and BRG1 or β-catenin in human cells [78], but the contribution of hTERT to the prevention of β-catenin nuclear retention and degradation was confirmed [79]. Similar observations led to a conclusion that potential influence of hTERT on stem cells might reflect two different pathways: hTERT could induce the transcription of β-catenin-dependent genes, or Wnt/β-catenin could directly affect the transcription of hTERT [80]. Since both, the Wnt pathway and telomerase, are engaged in the embryonic development and tissue homeostasis, a link between those two factors is suggested. In fact, transient activation of the Wnt/β-catenin pathway was shown to induce hTERT mRNA expression and elevate telomerase activity in different cell lines [81].
Additionally, a significant decrease of hTERT expression accompanied by telomerase inhibition and telomere attrition was noticed after silencing endogenous β-catenin [82]. Interestingly, hTERT contributed to modulate the expression of Wnt target genes, which play a role in the development and tumorigenesis [14, 15, 77, 83]. Surprisingly, telomerase role in the protection of mitochondrial integrity was also reported [84] as well as the contribution to NFkB signaling pathway [14, 15, 77, 83] which is activated by Tumor Necrosis Factor-α (TNF-α), endogenous ROS inducer [69]. Similarly, Mattiussi et al. reported that human telomerase was able to repress ROS-dependent intracellular signaling and TNF-α-mediated gene expression. Additionally, hTERT overexpression was reported to induce a strong constitutive nuclear accumulation of NFkBp65 [85]. Interestingly, it was also shown that a panel of genes was regulated by telomerase in acute myelogenous leukemia (AML) [86]. Blackburn et al., showed that in yeast cells after telomerase knockdown, alterations similar to a DNA damage responses and the environmental stress response appeared and it was accompanied by up-regulation of energy production genes and proliferation of mitochondria [87].
Altogether, the contribution of telomerase to the regulation of the expression of different genes is a well-known fact, and it was already thoroughly reviewed [87]. There is a pretty straight forward conclusion that ectopic expression of hTERT in human mammary epithelial cells correlates with the upregulation of growth-promoting genes, and the downregulation of growth inhibitory genes [87]. Thus, it is suggested that telomerase might interact with transcription factors or chromatin modifying factors that directly regulate certain gene transcription programs (e.g., NFkB p65 and β-catenin-dependent), and mediate the transcriptional regulation of target genes expression, however, the pool of those genes remains mostly unknown.
TERC in the regulation of gene expression
TERT is not the only one telomerase subunit that is capable of controlling the expression of human genes. It was shown in an animal model that suppression of mTERC in the murine melanoma B16 cell line provoked a significant downregulation of 138 genes. 8 of those genes were involved in the glycolytic pathway which suggests a functional association between telomerase and cancer metabolism [88] confirming an earlier report on this issue [89]. Blackburn’s group demonstrated that targeting hTERC in human colon cancer HCT116 cell line resulted in the suppression of angiogenesis and metastasis-associated genes. Some of these down-regulated genes, including Cyclin G2 and Cdc27, are involved in cell cycle progression. It became obvious that hTERC could also contribute to telomerase-mediated transcription of NFkB. Similarly, NFkB target genes were also reported downregulated in both, mTerc and mTert knockout mice, suggesting a feed-forward regulation between those factors [15]. Unfortunately, the concrete mechanism by which telomerase participates in the regulation of NFkB target genes has not been fully understood yet.
Telomerase as a target in immunotherapy
Last but not least, it seems that the non-canonical function of telomerase goes beyond all those mentioned pathways. Due to its key role in cancer metabolism but also a highly specific expression and activity, almost exclusively limited to cancer cells, telomerase seems to be a perfect target for immunotherapy. This approach includes two main trends in telomerase targeting in cancer treatment, i.e., anti-telomerase vaccines and the transfer of hTERT-specific cytotoxic T lymphocytes. Both those strategies were comprehensively presented in some latest works [90, 91].
Summary
Telomerase belongs to a narrow group of relatively specific markers that are promising targets in cancer therapy. But on the other hand, due to a well-known pro-survival activity of this enzyme, it is also considered as a potential tool in preventative treatments for neurodegenerative diseases, reducing disease symptoms or even extending lifespan as demonstrated in a mouse model of amyotrophic lateral sclerosis [57, 59]. There are still some concerns about the possible restoration of telomerase in normal (nonmalignant) cells since the immortality enzyme is suspected of triggering cancer development. However, those reports are ambiguous.
Importantly, telomerase is associated with so many telomere-unrelated functions that total elimination of this enzyme might bring a potential risk of unpredictable and severe side effects. It is actually known that overexpression of hTERT is correlated with advanced invasive stage of tumor progression and poor prognosis, but we cannot verify whether telomerase renewal is a reason or a consequence of cancer development. But the question is whether the same mechanisms rule cancer as well as normal cells and if these processes can be controlled. As demonstrated, hTERT may promote cancer and stem cell survival by lowering ROS levels. This would be beneficial in normal cells of course. Altogether it is not about questioning if hTERT should be turned off but rather if we can shut it down in target/cancer cells only?
References
Deng Y, Chang S (2007) Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab Investig 87:1071–1076
Bachand F, Autexier C (2001) Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA–protein interactions. Mol Cell Biol 21:1888–1897
Cong Y-S, Wright WE, Shay JW (2002) Human telomerase and its regulation. Microbiol Mol Biol Rev 66:407–425
De Vitis M, Berardinelli F, Sgura A (2018) Telomere length maintenance in cancer: at the crossroad between telomerase and alternative lengthening of telomeres (ALT). Int J Mol Sci 19:E606
Shay JW, Wright WE (2010) Telomeres and telomerase in normal and cancer stem cells. FEBS Lett 584:3819–3825
Calado RT, Young NS (2009) Mechanisms of disease: telomere diseases. N Engl J Med 361:2353–23652308
Davoli T, Denchi EL, de Lange T (2010) Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 141:81–93
Morrish TA, Greider CW (2009) Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet 5:e1000357
Murnane JP (2011) Telomere dysfunction and chromosome instability. Mutat Res 730(1–2):28–36. https://doi.org/10.1016/j.mrfmmm.2011.04.008
Rampazzo E, Bertorelle R et al (2010) Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br J Cancer 102:1300–1305
Oztas E, Kara H, Kara ZP et al (2016) Association between human telomerase reverse transcriptase gene variations and risk of developing breast cancer. Genet Test Mol Biomark 20:459–464
Zhang Y, Calado R, Rao M et al (2014) Telomerase variant A279T induces telomere dysfunction and inhibits non-canonical telomerase activity in esophageal carcinomas. PLoS ONE 9:e101010
Teichroeb JH, Kim J, Betts DH (2016) The role of telomeres and telomerase reverse transcriptase isoforms in pluripotency induction and maintenance. RNA Biol 13:707–719
Choi J, Southworth LK, Sarin KY et al (2008) TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet 4:e10
Ghosh A, Saginc G, Leow SC et al (2012) Telomerase directly regulates NF-kB-dependent transcription. Nat Cell Biol 14:1270–1281
Haendeler J, Dröse S, Büchner N et al (2009) Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol 29:929–935
Alcaraz-Pérez F, García-Castillo J, García-Moreno D et al (2014) A non-canonical function of telomerase RNA in the regulation of developmental myelopoiesis in zebrafish. Nat Commun 5:3228
Venkatesan RN, Price C (1998) Telomerase expression in chickens: constitutive activity in somatic tissues and down-regulation in culture. Proc Natl Acad Sci USA 95:14763–14768
Wege H, Müller A, Müller L et al (2007) Regeneration in pig livers by compensatory hyperplasia induces high levels of telomerase activity. Comp Hepatol 6:6
Nasir L, Devlin P, Mckevitt T et al (2001) Telomere lengths and telomerase activity in dog tissues: a potential model system to study human telomere and telomerase biology. Neoplasia 3:351–359
McClintock B (1941) The stability of broken ends of chromosomes in Zea Mays. Genetics 26:234–282
Chang S (2005) Modeling aging and cancer in the telomerase knockout mouse. Mutat Res 576:39–53
Bernardes de Jesus B, Vera E, Schneeberger K et al (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med 4:691–704
Allsopp RC, Vaziri H, Patterson C et al (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 89:10114–10118
Prowse KR, Greider CW (1995) Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci USA 92:4818–4822
Kirkpatrick KL, Clark G, Ghilchick M et al (2003) hTERT mRNA expression correlates with telomerase activity in human breast cancer. Eur J Surg Oncol 29:321–326
Rudolph KL, Chang S, Lee HW et al (1999) Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96:701–712
Xie Z, Jay KA, Smith DL et al (2015) Early telomerase inactivation accelerates aging independently of telomere length. Cell 160:928–939
Tong AS, Stern JL, Sfeir A et al (2015) ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep 13:1633–1646
Burchett KM, Etekpo A, Batra SK et al (2017) Inhibitors of telomerase and poly(ADP-ribose) polymerases synergize to limit the lifespan of pancreatic cancer cells. Oncotarget 8:83754–83767
Jafri MA, Ansari SA, Alqahtani MH, Shay JW (2016) Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med 8:69
Wanat JJ, Johnson FB (2012) Telomere stability and carcinogenesis: an off-again, on-again relationship. J Clin Invest 122:1962–1965
Huang FW, Hodis E, Xu MJ et al (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959
Maida Y, Kyo S, Kanaya T et al (2002) Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene 21:4071–4079
Liu X, Wang Y, Chang G et al (2017) Alternative splicing of hTERT pre-mRNA: a potential strategy for the regulation of telomerase activity. Int J Mol Sci 18(3):E567
Bojovic B, Booth RE, Jin Y et al (2015) Alternative lengthening of telomeres in cancer stem cells in vivo. Oncogene 34:611–620
Hahn WC, Counter CM, Lundberg AS et al (1999) Creation of human tumour cells with defined genetic elements. Nature 400:464–468
Perrem K, Colgin LM, Neumann AA et al (2001) Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol Cell Biol 21:3862–3875
Chang S, DePinho RA (2002) Telomerase extracurricular activities. Proc Natl Acad Sci USA 99:12520–12522
Cesare AJ, Kaul Z, Cohen SB et al (2009) Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol 16:1244–1251
Ford LP, Zou Y, Pongracz K et al (2001) Telomerase can inhibit the recombination-based pathway of telomere maintenance in human cells. J Biol Chem 276:32198–32203
Nakabayashi K, Ogata T, Fujii M et al (1997) Decrease in amplified telomeric sequences and induction of senescence markers by introduction of human chromosome 7 or its segments in SUSM-1. Exp Cell Res 235:345–353
Oh H, Taffet GE, Youker KA et al (2001) Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci USA 98:10308–10313
Artandi SE, Alson S, Tietze MK et al (2002) Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci USA 99:8191–8196
Stampfer MR, Garbe J, Levine G et al (2001) Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor beta growth inhibition in p16INK4A(−) human mammary epithelial cells. Proc Natl Acad Sci USA 98:4498–4503
Mukherjee S, Firpo EJ, Wang Y, Roberts JM (2011) Separation of telomerase functions by reverse genetics. Proc Natl Acad Sci USA 108:E1363–E1371
Smith LL, Coller HA, Roberts JM (2003) Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol 5:474–479
Castorina A, Szychlinska MA, Marzagalli R, Musumeci G (2015) Mesenchymal stem cells-based therapy as a potential treatment in neurodegenerative disorders: is the escape from senescence an answer? Neural Regen Res 10:850–858
Coluzzi E, Colamartino M, Cozzi R et al (2014) Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 9:e110963
Grollman AP, Moriya M (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet 9:246–249
Gordon DM, Santos JH (2010) The emerging role of telomerase reverse transcriptase in mitochondrial DNA metabolism. J Nucleic Acids. https://doi.org/10.4061/2010/390791
Miao G-Y, Zhou X, Zhang X et al (2016) Telomere-mitochondrion links contribute to induction of senescence in MCF-7 cells after carbon-ion irradiation. Asian Pac J Cancer Prev 17:1993–1998
Singhapol C, Pal D, Czapiewski R et al (2013) Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE 8:e52989
Muzza M, Colombo C, Cirello V et al (2016) Oxidative stress and the subcellular localization of the telomerase reverse transcriptase (TERT) in papillary thyroid cancer. Mol Cell Endocrinol 431:54–61
Kovalenko OA, Caron MJ, Ulema P et al (2010) A mutant telomerase defective in nuclear-cytoplasmic shuttling fails to immortalize cells and is associated with mitochondrial dysfunction. Aging Cell 9:203–219
Ling X, Wen L, Zhou Y (2012) Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. Int J Med Sci 9:545–554
Bollmann FM (2013) Telomerase inhibition may contribute to accelerated mitochondrial aging induced by anti-retroviral HIV treatment. Med Hypotheses 81:285–287
Yan J, Zhou Y, Chen D et al (2015) Impact of mitochondrial telomerase over-expression on drug resistance of hepatocellular carcinoma. Am J Transl Res 7:88–99
Zhang P, Pan H, Wang J et al (2014) Telomerase activity-independent function of telomerase reverse transcriptase is involved in acrylamide-induced neuron damage. Biotech Histochem 89:327–335
Park H-H, Lee K-Y, Kim S et al (2014) Novel vaccine peptide GV1001 effectively blocks β-amyloid toxicity by mimicking the extra-telomeric functions of human telomerase reverse transcriptase. Neurobiol Aging 35:1255–1274
Li P, Tong Y, Yang H et al (2014) Mitochondrial translocation of human telomerase reverse transcriptase in cord blood mononuclear cells of newborns with gestational diabetes mellitus mothers. Diabetes Res Clin Pract 103:310–318
González-Giraldo Y, Forero DA, Echeverria V et al (2016) Neuroprotective effects of the catalytic subunit of telomerase: a potential therapeutic target in the central nervous system. Ageing Res Rev 28:37–45
Spilsbury A, Miwa S, Attems J, Saretzki G (2015) The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J Neurosci 35:1659–1674. https://doi.org/10.1523/JNEUROSCI.2925-14.2015
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12:609–622
Eitan E, Tichon A, Gazit A et al (2012) Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol Med 4:313–329
Vaváková M, Ďuračková Z, Trebatická J (2015) Markers of oxidative stress and neuroprogression in depression disorder. Oxid Med Cell Longev 2015:898393
Eitan E, Braverman C, Tichon A et al (2016) Excitotoxic and radiation stress increase TERT levels in the mitochondria and cytosol of cerebellar Purkinje neurons. Cerebellum 15:509–517
Gutkin A, Uziel O, Beery E et al (2016) Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 7:59173–59188
Liu N, Ding D, Hao W et al (2016) hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res 44:8693–8703
Falchetti ML, Mongiardi MP, Fiorenzo P et al (2008) Inhibition of telomerase in the endothelial cells disrupts tumor angiogenesis in glioblastoma xenografts. Int J Cancer 122:1236–1242
George J, Banik NL, Ray SK (2009) Combination of hTERT knockdown and IFN-gamma treatment inhibited angiogenesis and tumor progression in glioblastoma. Clin Cancer Res 15:7186–7195
Zhou L, Zheng D, Wang M, Cong Y-S (2009) Telomerase reverse transcriptase activates the expression of vascular endothelial growth factor independent of telomerase activity. Biochem Biophys Res Commun 386:739–743
Zheng X-H, Nie X, Liu H-Y et al (2016) TMPyP4 promotes cancer cell migration at low doses, but induces cell death at high doses. Sci Rep 6:26592. https://doi.org/10.1038/srep26592
Liu H, Liu Q, Ge Y et al (2016) hTERT promotes cell adhesion and migration independent of telomerase activity. Sci Rep 6:22886
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Park J-I, Venteicher AS, Hong JY et al (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460:66–72
Listerman I, Gazzaniga FS, Blackburn EH (2013) An investigation of the effects of the telomerase core protein TERT on Wnt signaling in human breast cancer cells. Mol Cell Biol 34(2):280–289
Liu Z, Li Q, Li K et al (2013) Telomerase reverse transcriptase promotes epithelial–mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 32:4203–4213
Hoffmeyer K, Raggioli A, Rudloff S et al (2012) Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336:1549–1554
Lafferty-Whyte K, Cairney CJ, Will MB et al (2009) A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 28:3765–3774
Zhang Y, Toh L, Lau P, Wang X (2012) Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J Biol Chem 287:32494–32511
Ding D, Xi P, Zhou J et al (2013) Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-kB-dependent transcription. FASEB J 27:4375–4383
Chiodi I, Mondello C (2012) Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front Oncol 2:133
Mattiussi M, Tilman G, Lenglez S, Decottignies A (2012) Human telomerase represses ROS-dependent cellular responses to Tumor Necrosis Factor-α without affecting NF-kB activation. Cell Signal 24:708–717
Bagger FO, Bruedigam C, Lane SW (2016) Analysis of telomerase target gene expression effects from murine models in patient cohorts by homology translation and random survival forest modeling. Genomics Data 7:275–280
Nautiyal S, DeRisi JL, Blackburn EH (2002) The genome-wide expression response to telomerase deletion in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 99:9316–9321
Bagheri S, Nosrati M, Li S et al (2006) Genes and pathways downstream of telomerase in melanoma metastasis. Proc Natl Acad Sci USA 103:11306–11311
Li S, Crothers J, Haqq CM, Blackburn EH (2005) Cellular and gene expression responses involved in the rapid growth inhibition of human cancer cells by RNA interference-mediated depletion of telomerase RNA. J Biol Chem 280:23709–23717
Kailashiya C, Sharma HB, Kailashiya J (2017) Telomerase based anticancer immunotherapy and vaccines approaches. Vaccine 35:5768–5775
Carrozza F, Santoni M, Piva F, Cheng L, Lopez-Beltran A, Scarpelli M, Montironi R, Battelli N, Tamberi S (2018) Emerging immunotherapeutic strategies targeting telomerases in genitourinary tumors. Crit Rev Oncol Hematol 131:1–6
Acknowledgements
The present review was supported by a grant from the National Science Centre: 2016/21/B/NZ7/01079.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Romaniuk, A., Paszel-Jaworska, A., Totoń, E. et al. The non-canonical functions of telomerase: to turn off or not to turn off. Mol Biol Rep 46, 1401–1411 (2019). https://doi.org/10.1007/s11033-018-4496-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4496-x