
Abstract
Valorisation of tuber protein is relevant for the potato starch industry to create added-value and reduce impact on the environment. Hence, protein content has emerged as a key quality trait for innovative potato breeders. In this study, we estimated trait heritability, explored the relationship between protein content and tuber under-water weight (UWW), inferred haplotypes underlying quantitative trait loci (QTLs) and pinpointed candidate genes. We used a panel of varieties (N = 277) that was genotyped using the SolSTW 20 K Infinium single-nucleotide polymorphism (SNP) marker array. Protein content data were collected from multiple environments and years. Our genome-wide association study (GWAS) identified QTLs on chromosomes 3, 5, 7 and 12. Alleles of StCDF1 (maturity) were associated with QTLs found on chromosome 5. The QTLs on chromosomes 7 and 12 are presented here for the first time, whereas those on chromosomes 3 and 5 co-localized with loci reported in earlier studies. The candidate genes underlying the QTLs proposed here are relevant for functional studies. This study provides resources for genomics-enabled breeding for protein content in potato.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Global population growth, accompanied with increased consumer wealth, will change food consumption patterns worldwide (Tilman and Clark 2014). By the year 2050, the projected demand for protein from animal sources is expected to double from 2000 (Alexandratos 1999). This trend raises sustainability and food security concerns, as the intensive production of animal protein adds pressure on the environment—as vast amounts scarce (non-renewable) resources such as land, water and minerals are needed. On the contrary, the production of plant protein is more sustainable for the environment as less resources are needed (Sabaté and Soret 2014).
Potato (Solanum tuberosum L.) is a well-known starch crop. However, few realise that the potato crop also serves as an abundant source of plant protein (Jørgensen et al. 2006). Although protein content in potato tubers is relatively low (0.32–1.63%) (Bárta et al. 2012; Klaassen et al. 2019; Ortiz-Medina 2006), protein yield per hectare (ha) is eminent due to the high-yielding ability and high harvest-index of the potato crop that can reach up to 124 ton−1ha (Kunkel and Campbell 1987). The potato starch industry processes potatoes to produce starch and by-products. After starch is extracted from tubers, potato fruit juice (PFJ) is released as a major aqueous by-product that contains protein. After proteins are extracted from PFJ, functional (native) potato protein isolates may be utilized in high-end food and pharmaceutical applications that include foaming agents, anti-oxidants, emulsifiers (Creusot et al. 2011; Edens et al. 1999; Kudo et al. 2009), inhibitors of faecal proteolytic compounds that cause dermatitis (Ruseler-van Embden et al. 2004) and satiety agents (Hill et al. 1990). Therefore, valorisation of protein provides opportunities to create added-value for the potato starch industry. Consequently, innovative firms in the industry are keen to use protein-rich potato varieties and therefore high protein content has emerged as a key quality trait for breeders. However, breeding for protein content in potato is challenging due to the complex genetic basis underlying the trait (Klaassen et al. 2019). To facilitate breeding for protein content in potato, improved comprehension of the inheritance, quantitative trait loci (QTLs) and relationships with other agronomical relevant traits are useful.
Knowledge on the inheritance of protein content in potato is limited. To the best of our knowledge, three genetic studies on protein content in bi-parental populations have been published (Acharjee et al. 2018; Klaassen et al. 2019; Werij 2011). These studies estimated moderate levels of trait heritability (40–74%) and identified minor-effect QTLs on chromosomes 1, 2, 3, 5 and 9 in both non-cultivated diploid and cultivated tetraploid potato germplasm. As for other crops that include soybean, maize and wheat (Balyan et al. 2013; Hwang et al. 2014; Karn et al. 2017), protein content has been described as a complex trait that is regulated by a plethora of interactions between genetic and environmental factors. Therefore, QTLs for protein content in heterozygous tetraploid (2n = 4x = 48) potato are likely to be affected by both epistasis and environmental factors.
Genome-wide association studies (GWAS) have been used as a method to dissect the genetic architecture of complex traits in multiple species that include potato (Rosyara et al. 2016; Sharma et al. 2018). As opposed to genetic studies performed on bi-parental populations, GWAS offers the advantage to identify QTLs within a panel of diverse individuals, and to potentially gain a high mapping resolution for identifying candidate genes.
In this study, we carried out a GWAS to dissect the genetics of protein content in a panel of tetraploid potato. We report on the relationship between protein content and tuber under-water weight (a proxy for starch content), haplotypes underlying QTLs and putative candidate genes.
Materials and methods
Germplasm collection
The panel (N = 277) consisted of tetraploid (2n = 4x = 48) individuals. The panel was composed of 189 varieties (D’hoop et al. 2008) and 88 starch potato progenitors that originated from five potato breeding companies (Agrico, Averis Seeds, C. Meijer, HZPC and KWS) (Supplementary Table 1). These included both modern and old individuals from different market segments and geographic origins. Analysis of population structure in the panel displayed three sub-populations, as reported earlier (D’hoop et al. 2008; Vos 2016). These sub-populations, hereafter referred to as “Processing”, “Other” and “Starch”, were used for analyses.
Field trials
Raw phenotypic data were collected over years and locations (multi-location, multi-year) from unbalanced field trials that were carried out in the Netherlands. These trials were carried out in years 2008–2010 in Bant, Emmeloord, Metslawier, Rilland and Valthermond. The accessions were replicated three times or more, except for nine accessions that were replicated twice or once. A replicate (experimental unit) consisted of a four-plant plot within a row in the field. Raw phenotypic data were used to compute the BLUEs for the accessions. The trials were carried out during the conventional potato growing seasons in the Netherlands as described by D’hoop et al. (2008). Uniform seed tubers were used as planting material and were propagated at a single location 1 year prior to the trials. The seed potatoes were planted at 75-cm spacing between the rows and 35 cm between the hills. Guard rows were used to separate the plots in the trial. Regular husbandry practices for potato production in the Netherlands were carried out during the field trials. After harvest, the tubers were stored under cool conditions prior to use.
Quantification of phenotypes
Soluble protein content in potato fruit juice (PFJ) was determined by using the bicinchoninic acid (BCA) assay (Smith et al. 1985). Bovine serum albumin (BSA) was used as a standard. Protein content was quantified as described by Klaassen et al. (2019). Tuber under-water weight (UWW), a proxy for starch content, was quantified as described in a previous study (Bradshaw et al. 2008).
Best linear unbiased estimates
To estimate the best linear unbiased estimates (BLUEs), a mixed model was used. BLUEs were computed using restricted maximum likelihood (REML) (D’hoop et al. 2011) as follows:
where the “μ” represented the overall mean response, the “Accession” term was fixed. The “Year”, “Location” and residual “Error” terms were random. Broad sense heritability estimates (H2) were computed from variance components (see also D’hoop et al. 2011) as follows:
where the variance components σ2G (“Accession”), σ2G × Y (“Accession” × “Year”), σ2G × L (“Accession” × “Location”), σ2G × Y × L (“Accession” × “Year” × “Location”) and σ2e (residual “Error”) were derived from REML. In a second REML model, the “Accession” term was fixed. In the H2 equation, the terms “y”, “l” and “r” represented the number of years, number of locations and number of biological replicates respectively.
Genotyping and genotype calling
The panel was genotyped using the SolSTW 20 K Infinium SNP marker array (Vos et al. 2015). Genotype calling (assignment of SNP allele dosages) were carried out by using fitTetra (Voorrips et al. 2011) and Illumina GenomeStudio software version 2010.3 (Illumina, San Diego, CA, USA), as described by Vos et al. (2015). The threshold for minor-allele frequency (MAF) was set at 1.5% (equivalent to 6% for tetraploid potato with four sets of homologous chromosomes). After filtering, 14,436 high-quality SNP markers were used for GWAS. The physical coordinates of the SNPs were based on the potato reference genome, i.e. pseudomolecules v4.03 (PGSC 2011).
Population structure analysis
The population structure of the panel was analysed by using STRUCTURE software package v2.3.4 (Pritchard et al. 2000). Ten runs were performed to estimate the K values using 2000 randomly selected SNPs. A Markov chain Monte Carlo (MCMC) burn-in period of 10,000 was used and the number of iterations was set at 10,000. The appropriate number of sub-populations were determined from delta K and optimal K values (Evanno et al. 2005) based on output data derived from STRUCTURE Harvester (Earl and vonHoldt 2012) (http://taylor0.biology.ucla.edu/structureHarvester/). Membership probability estimates from thirty runs were averaged and used to assign each individual to cluster groups (sub-populations). The sub-populations were denoted as “Processing”, “Other” and “Starch”, based on prior knowledge that these three sub-populations existed in the panel (D’hoop et al. 2008; Vos 2016).
Genome-wide association study
A GWAS was performed using the phenotype (BLUEs) and genotype data. A naive model, a mixed model and a conditional mixed model were used to compute associations. For the naive model, associations between the BLUEs and SNP dosages were analysed. The mixed model was used to perform association analysis whilst correcting for kinship (K). As population structure was weak for the panel, as shown by (Vos et al. 2017), we did not correct for sub-populations (Q). For the mixed model, we used the same SNPs for GWAS and kinship correction. A sub-set of these SNPs was used for inference of population structure. To dissect the effect of maturity alleles (StCDF1) that are physically positioned at the start of chromosome 5, a conditional mixed model was used. The conditional mixed model, as described in earlier studies (Kang et al. 2010; Segura et al. 2012), included the SNP marker “PotVar0079081” (Chromosome 5, coordinate 4,489,481 Mbp) as a cofactor that tagged the early maturity allele (StCDF1.1) in a haplotype-specific manner (Willemsen 2018). The following equations were used for the GWAS models:
In the equations 3, 4 and 5, “Y” represents the BLUEs, “X” represents the SNP markers (fixed effect), “K” represents the random kinship (co-ancestry) matrix and “A” represents the SNP marker set as cofactor (fixed). The term “ε” represents the vector of random residual errors. The term “α” represents the estimated SNP effects, “β” represents the estimated effect of the SNP marker set as cofactor and “μ” represents the estimated kinship variance component. Analyses were performed in R software package GWASpoly (Rosyara et al. 2016). Phenotypic variance explained (R2) by SNPs were calculated from squared correlation coefficients between the BLUEs and SNP dosage scores (allele copy number).
Significance threshold and QTL support interval
Manhattan plots were used to illustrate the genome-wide association scores of SNPs. These scores were computed from P values of the SNPs as follows:
We used several significance thresholds to identify QTLs. To correct for multiple testing, we used the 5% Bonferroni threshold (−log10(P) = 5.3). The Bonferroni threshold is known to inflate the probability of Type II errors (false-negative findings) in the presence of high linkage disequilibrium between markers (Gao et al. 2008; Johnson et al. 2010). Therefore, the 5% Li and Ji threshold was also computed by correlated multiple testing (Li and Ji 2005) (−log10(P) = 3.9). Correlated multiple testing was conducted at α = 0.05, to adjust for the effective number of independent tests and compensate for Type II errors. For naive analyses, permutation testing was carried out with N = 1000 permutations at α = 0.05 to define the threshold value (−log10(P) = 5.0) (Churchill and Doerge 1994) (−log10(P) = 5.0). The support intervals of QTLs were set at 1.5 Mbp for non-introgressed regions and 2.5 Mbp for introgressed regions as described by Vos (2016).
Haplotype inference
Determination of haplotypes underlying QTLs was performed using a contemporary haplotype inference method developed in tetraploid potato (Willemsen 2018). This method estimated the linkage phase between pairs of SNP markers, followed by joining of linked SNPs into haplotypes. Only SNPs exceeding the Li and Ji threshold (−log10(P) = 3.9) were used for haplotype construction and to obtain the dosages of the haplotypes.
SNP allele frequency
The SNP allele frequency (%) in the panel of tetraploid accessions was computed by using the SNP dosage scores and number of accessions as follows:
Results
Phenotypic variation of the traits (BLUEs)
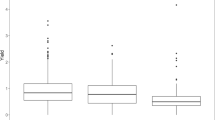
The panel displayed variation for the BLUEs of protein content, tuber under-water weight (UWW) and protein content in potato fruit juice (PFJ) (Fig. 1). Protein content ranged between 0.73–1.72% (w/w). The broad sense heritability estimates (H2) for protein content, protein content in PFJ and UWW were 48%, 58% and 81% respectively (Table 1). BLUEs for UWW and protein content in PFJ ranged between 270 and 572 g 5 kg−1 and 0.89–2.44% (w/v) respectively. To evaluate correlations between the traits, scatterplots for the phenotypic values (BLUEs) were evaluated. Moderate to high correlations (P < 0.001) were observed for the phenotypic BLUEs (Fig. 1) (protein content versus UWW: r = 0.639; UWW versus protein content PFJ: r = 0.746; protein content versus protein content PFJ: r = 0.988).

Distributions and scatterplots of the phenotypic values (BLUEs). Distributions for a protein content (% w/w), b tuber under-water weight (UWW) (g 5 kg−1) and c protein content PFJ (potato fruit juice PFJ) (% w/v). Scatter plots for d protein content versus under-water weight (UWW), e UWW versus protein content PFJ (potato fruit juice) and f protein content versus protein content PFJ. The linear regression lines are shown in blue. SD,standard deviation
Population structure
Population structure of the panel was analysed using SNP marker data from the array. Three clusters (sub-populations) were characterized (Supplementary Fig. 2) and were denoted as “Processing” (N = 35), “Other” (N = 136) and “Starch” (N = 106). As the sub-populations showed unequal trait values (One-way ANOVA, P = 2.46 × 10−3; Supplementary Fig. 1), protein content was found to be confounded with population structure. Likewise, the Q-Q plot for naive GWAS on the panel showed inflated probabilities (Supplementary Fig. 3), that may have been caused by population structure. Therefore, a kinship-corrected GWAS was also performed to identify QTLs for protein content (Fig. 2).

Manhattan plot for kinship-corrected GWAS on the panel (N = 277) with Quantile-Quantile plot for the observed versus expected probabilities. Top (red) line indicates Bonferroni threshold at 5.3. Lower (blue) line indicates Li and Ji threshold at 3.9
Identification of QTLs
To identify QTLs for protein content, a kinship-corrected GWAS was carried out with 14,436 SNPs using the panel of 277 accessions. Three QTLs were identified above the Li and Ji threshold (−log10(P) = 3.9) on chromosomes 3, 5 and 7 (Fig. 2), that each explained 9–12% of the phenotypic variance (R2) (Table 2). The strongest association, that also exceeded the Bonferroni threshold, was found at the start of chromosome 5 at 4.71 Mbp (−log10(P) = 5.84; R2 = 0.11). The end of chromosome 3 harboured a QTL at 60.84 Mbp (−log10(P) = 4.07; R2 = 0.11). A third QTL was positioned at the end of chromosome 7 at 50.15 Mbp (−log10(P) = 3.97; R2 = 0.12). The naive GWAS identified significant QTLs on all the twelve potato chromosomes (Supplementary Fig. 3), but were expected to be false-positive associations because the trait values were confounded with population structure in the panel (Supplementary Fig. 1). To dissect the potential year (season) effects, correlation analysis and GWAS were performed on the BLUEs for 2008, 2009 and 2019 (Supplementary Figs. 7 and 8). As observed for the panel, GWAS on the BLUEs for 2008 showed associations at the start of chromosome 5. The BLUEs for 2009 produced associations again at the start of chromosome 5 and at the end of chromosome 7. For the BLUEs of 2010, associations were found at the ends of chromosomes 2 and 11. Moderate to high correlations were observed for the BLUEs between the individual years. As the raw values for UWW and protein content in PFJ were used to correct for the values for protein content, GWAS were performed on these two traits as well (Supplementary Fig. 6).
To pinpoint putative candidate genes from the genomic regions underlying the QTLs, linkage disequilibrium (LD)-based QTL support intervals were used as described by Vos et al. (2017). The genes underlying these intervals were retrieved from the potato reference genome (PGSC 2011). From the longlists of genes (Supplementary Table 4), putative candidates were selected based on their annotation (gene name). As a result, the QTL interval on chromosome 3 co-localized with a nitrate transporter (60.09 Mbp). The interval on chromosome 5 harboured StCDF1 (4.54 Mbp) and a cluster of nine nitrate transporters (6.00–7.52 Mbp). No obvious candidate genes could be proposed to be implicated with the QTL on chromosome 7.
Haplotypes underlying QTLs
A contemporary approach by Willemsen (2018) was used to determine the haplotype-specificity of the SNP markers underlying the QTLs. Results showed that all SNPs underlying the QTL on chromosome 5 (that exceeded the Li and Ji threshold) were haplotype-specific (Table 2). These SNPs were haplotype-specific for a late maturity allele of StCDF1 (Supplementary Table 2), as proposed by Willemsen (2018). Moreover, these SNPs also tagged a unique introgression segment from wild potato (Solanum vernei Bitter & Wittm.) as described by van Eck et al. (2017). Over the years, this introgression segment has been used by potato breeders to introduce resistance against Globodera pallida nematodes (the so-called Gpa5 locus) in the genepool of cultivated potato (Rouppe van der Voort et al. 2000; Van Eck et al. 2017). Graphical genotypes of the panel, as performed by van Eck et al. (2017), illustrated that this introgression segment was mainly present in the starch varieties and starch progenitors. For these varieties and progenitors, the introgression segment was found to be present in either simplex (a single copy) or duplex (two copies) form (Supplementary Fig. 4). The SNPs underlying the QTLs on chromosome 3 and 7 were not found to be haplotype-specific.
Variance explained by multiple QTLs
By using multiple linear regression, we tested the cumulative effect of multiple significant SNP markers underlying QTLs together. The SNPs underlying the QTLs on chromosomes 3, 5 and 7 together explained 22% of the variance (Supplementary Table 3). When the SNP on chromosome 5 was excluded, the QTLs on chromosomes 3 and 7 together explained 21%. The combination of SNPs on chromosomes 5 and 7 jointly explained 20%. The QTLs on chromosomes 3 and 5 jointly explained less variance (13%).
Sub-population QTLs
In an attempt to circumvent the confounding effect of population structure in the panel, GWAS was performed on the sub-populations “Starch” (N = 106) and “Other” (N = 136). The sub-population “Processing” was not included as it consisted of a relatively small number of individuals (N = 35). Kinship-corrected GWAS on the sub-population “Starch” identified one QTL above the Li and Ji threshold at the end of chromosome 3 (R2 = 0.15) (Fig. 3; Table 3). The QTL peak caused by SNP marker PotVar0020225 was positioned 0.708 Mbp north from the QTL identified in the panel (Table 2). The naive GWAS on the sub-population “Starch” did not identify significant QTLs, although noticeable associations were found slightly below the thresholds at the end of chromosome 3 (Supplementary Fig. 5). Kinship-corrected GWAS on the sub-population “Other” identified a QTL at the start of chromosome 5 (R2 = 0.15) (Fig. 3). This sub-population QTL was positioned 0.975 Mbp (Table 3) south from the QTL found in the panel, that was introgressed from wild potato into the starch varieties and starch progenitors as a source of resistance against nematodes (Table 2). The SNPs tagging this haplotype in the sub-population “Other” were lower than the minor allele frequency (MAF) threshold of 1.5%. Therefore this haplotype remained unnoticed and could not uncover a QTL.

Manhattan plots for kinship-corrected GWAS on sub-populations a “Starch” (N = 106), b “Other” (N = 136) and c “Other” (N = 136) by including SNP marker “PotVar0079081” as a cofactor for early maturity (StCDF1.1). Quantile-Quantile plots for the observed versus expected probabilities are shown on the right. Top (red) line indicates Bonferroni threshold at 5.3. Lower (blue) line indicates Li and Ji threshold at 3.9
To verify whether or not the QTL at the start of chromosome 5 was associated with plant maturity (StCDF1) (Kloosterman et al. 2013), we performed conditional kinship-corrected GWAS on the sub-population “Other” by using the SNP marker “PotVar0079081” as a cofactor that tags the early maturity allele (StCDF1.1), as described by Willemsen (2018). This approach, reduced the significance of the original QTL at the start of chromosome 5 (from −log10(P) = 4.46 down to 3.19) (Fig. 3). This finding suggested that the maturity score of potato varieties, as largely controlled by StCDF1.1, indirectly influenced protein content in this sub-population. By performing the cofactor analysis, an otherwise masked QTL was uncovered at the end of chromosome 12 (Peak SNP: “PotVar0052807”; 59,294,858 bp; −log10(P) = 4.63). Naive GWAS on the sub-population “Other” showed inflated associations that probably caused false-positive QTLs on chromosomes 1, 2, 3, 4, 5, 7 and 10 (Supplementary Fig. 5).
Discussion
GWAS as a tool to detect QTLs
We used GWAS to shed light on the complex genetic architecture of protein content in potato. We identified QTLs with minor effects on chromosomes 3, 5, 7 and 12 (Fig. 2; Fig. 3). The QTLs identified on chromosomes 3 and 5, coincided with previous studies (Acharjee et al. 2018; Klaassen et al. 2019; Werij 2011). For chromosome 3, the QTL identified in the entire panel was also observed in the sub-population “Starch”. For chromosome 5, we uncovered an introgression segment from wild potato that was associated with protein content (Supplementary Fig. 4). This introgressed segment harboured a late maturity allele of StCDF1 (Supplementary Table 2), as well as the Gpa5 resistance allele against potato cyst nematodes (Globodera pallida). However, the SNPs tagging this introgression segment did not bring forth a QTL in the sub-population “Starch”, even though the allele frequency of these SNPs in this sub-population was considerable (9–10%). We also observed that the additive effect of this QTL was lower than expected when combined with the other two QTLs on chromosomes 3 and 7 (Supplementary Table 3). We showed that protein content was confounded with population structure in the panel. This result was likely caused by higher BLUEs values for protein content in the sub-population “Starch” (Supplementary Fig. 1). Therefore, we propose that the QTL on chromosome 5 in the panel could be an artefact. Validation studies, for instance using bi-parental mapping populations, may confirm the relevance of SNPs underlying this QTL for use in breeding to improve protein content. If these SNPs are to be used for breeding, they will at least provide a source of resistance against cyst nematodes and contribute towards a later maturity index due to StCDF1. In the sub-population “Other” we also identified a QTL at the start of chromosome 5. Conditional GWAS on this sub-population showed that this association was not caused by the introgression segment from wild potato. Instead, this QTL coincided with the early maturity allele of StCDF1 (StCDF1.1). Findings from GWAS on the panel as well as the sub-populations showed that different haplotypes at the start of chromosome 5 were associated with protein content.
To the best of our knowledge, the identified QTLs on chromosomes 7 and 12 have not been described before in literature. Bi-parental populations, that descend from crosses between protein-rich varieties, can be used to test/validate and stack multiple copies of favourable variants/alleles for multiple protein content QTLs simultaneously. For instance, the cross between the starch varieties Kartel × Seresta will allow the SNPs underlying all three QTLs identified in the panel here, to segregate in nulliplex (null), simplex (one) and duplex (two) dosages in the F1 progeny. This cross will provide improved insight into the cumulative effects of the underlying haplotypes. Our results, as presented in Supplementary Table 3, suggest both additive and epistatic effects of the SNPs (alleles). We observed that the effects of genotype-by-environment (G × E) interactions were small to moderate for protein content (Table 1). On the other hand, a large proportion of variance was ascribed to the residuals (error). Hence, future genetic studies on protein content may be improved by reducing the residual error in these experiments.
Missing heritability
Studies in soybean, wheat and maize describe protein content as a complex trait that is governed by multiple genes and environmental factors. We estimated a moderate trait heritability for protein content (H2 = 0.48). This H2 value ranged between 40 and 74%, i.e. in line with previous studies (Klaassen et al. 2019; Werij 2011). GWAS on the panel identified three QTLs that cumulatively explained 22% of the variance. Hence, we demonstrate a clear example of missing heritability. Several factors may have contributed to this finding, that include the limited statistical power to detect loci with small effects, interactions between loci, effects or rare variants and potential banishment of true-positive QTLs due to kinship correction. Alternatively, overestimation of the broad sense heritability estimate (H2) may also have occurred. In any case, it should be noted that our H2 will be much larger than the narrow sense (h2) estimate.
To optimize the detection of QTLs by GWAS, the design and methodology should be considered carefully. Using more individuals will likely increase statistical power, as shown in numerous human and crop genetic studies, e.g. for soybean (Bandillo et al. 2015). Optimization of GWAS will likely identify loci with minor effects or those caused by rare variants with a low allele frequency. Certainly the population structure, distribution of the phenotypic values, as well as the ascertainment bias of SNPs in marker arrays should be considered beforehand as proposed by Vos (2016).
Correlation between tuber protein content and under-water weight
For other crops, a negative correlation is often observed between protein content and other major (seed) storage compounds, e.g. oil content in soybean (Patil et al. 2017). Interestingly, while expecting a similar trade-off in potato, we found a moderate positive correlation (r = 0.64) between protein content and under-water weight (UWW: a proxy for starch content) (Fig. 1). Therefore, selection pressure for high UWW in the starch genepool, aimed to increase starch content, may have coincided with unconscious selection for high protein content (Supplementary Fig. 1). Kinship-corrected GWAS on UWW in the panel did not identify potential associations between UWW and maturity alleles of StCDF1 at the start of chromosome 5 (Supplementary Fig. 6). The statistical power produced by the 277 individuals here may have been insufficient to uncover significant signals due to the complex (polygenic) genetic architecture of starch content in potato. A positive correlation between protein content and UWW suggests that these traits may be (partly) interrelated due to shared biological mechanisms. It is well established that photosynthesis-derived carbon and nitrogen assimilation pathways are connected and tightly controlled in plants. Molecular studies have shown that intracellular glucose is used by plants to synthesize both protein and starch (Bihmidine et al. 2013). Reduced levels of ADP-glucose (i.e. glucosyl donor of glucose) by inactivated ADP-glucose pyrophosphorylase (AGPase) in barley mutants, was accompanied with the downregulation of genes related to amino acid and storage protein biosynthesis (Faix et al. 2012). Therefore, the genes that regulate protein content in potato may affect starch content, yet this point remains to be addressed in future studies. Unravelling the positive correlation between protein and starch content in potato, will certainly be dealt with in future studies.
Putative candidate genes for protein content
To pinpoint putative candidate genes, we used LD-bound QTL support intervals to narrow down on genomic regions. This approach identified several candidates that included StCDF1 (maturity) and nitrate transporters (Supplementary Table 4). Conditional GWAS on the sub-population “Other” showed that a late maturity allele of StCDF1 was positively associated with protein content. Nitrate transporters are known to function in the uptake and allocation of inorganic nitrate (NO3) in plants (Hsu and Tsay 2013; Léran et al. 2014). Nitrate is the predominant nitrogen-containing macronutrient in aerobic soils under temperate climatic conditions. Hence, allelic variants of nitrate transporters may differ in nitrate uptake and interaction with nitrogen-responsive genes that ultimately affect protein content, as proposed for rice (Hu et al. 2015). Future molecular studies on the above mentioned candidate genes that include gene expression, overexpression and knock-out studies, are certainly relevant to study their biological functions and effects on protein content in potato.
References
Acharjee A, Chibon P-Y, Kloosterman B, America T, Renaut J, Maliepaard C, Visser RG (2018) Genetical genomics of quality related traits in potato tubers using proteomics. BMC Plant Biol 18(1):20
Alexandratos N (1999) World food and agriculture: outlook for the medium and longer term. Proc Natl Acad Sci 96(11):5908–5914
Balyan HS, Gupta PK, Kumar S, Dhariwal R, Jaiswal V, Tyagi S, Agarwal P, Gahlaut V, Kumari S (2013) Genetic improvement of grain protein content and other health-related constituents of wheat grain. Plant Breed 132(5):446–457
Bandillo N, Jarquin D, Song Q, Nelson R, Cregan P, Specht J, Lorenz A (2015) A population structure and genome-wide association analysis on the USDA soybean germplasm collection. Plant Genome 8(3)
Bárta J, Bártová V, Zk Z, Šedo O (2012) Cultivar variability of patatin biochemical characteristics: table versus processing potatoes (Solanum tuberosum L.). J Agric Food Chem 60(17):4369–4378
Bihmidine S, Hunter C, Johns C, Koch K, Braun D (2013) Regulation of assimilate import into sink organs: update on molecular drivers of sink strength. Front Plant Sci 4(177). https://doi.org/10.3389/fpls.2013.00177
Bradshaw JE, Hackett CA, Pande B, Waugh R, Bryan GJ (2008) QTL mapping of yield, agronomic and quality traits in tetraploid potato (Solanum tuberosum subsp. tuberosum). Theor Appl Genet 116(2):193–211
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
Creusot N, Wierenga PA, Laus MC, Giuseppin ML, Gruppen H (2011) Rheological properties of patatin gels compared with β-lactoglobulin, ovalbumin, and glycinin. J Sci Food Agric 91(2):253–261
D’hoop BB, Paulo MJ, Mank RA, Van Eck HJ, Van Eeuwijk FA (2008) Association mapping of quality traits in potato (Solanum tuberosum L.). Euphytica 161(1–2):47–60
D’hoop BB, Paulo MJ, Visser RGF, Van Eck HJ, Van Eeuwijk FA (2011) Phenotypic Analyses of Multi-Environment Data for Two Diverse Tetraploid Potato Collections: Comparing an Academic Panel with an Industrial Panel. Potato Research 54(2):157–181
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Edens L, Plijter JJ, Van DLJAB (1999) Novel food compositions. Google Patents
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Faix B, Radchuk V, Nerlich A, Hümmer C, Radchuk R, Emery RN, Keller H, Götz KP, Weschke W, Geigenberger P (2012) Barley grains, deficient in cytosolic small subunit of ADP-glucose pyrophosphorylase, reveal coordinate adjustment of C: N metabolism mediated by an overlapping metabolic-hormonal control. Plant J 69(6):1077–1093
Gao X, Starmer J, Martin ER (2008) A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 32(4):361–369
Hill AJ, Peikin SR, Ryan CA, Blundell JE (1990) Oral administration of proteinase inhibitor II from potatoes reduces energy intake in man. Physiol Behav 48(2):241–246
Hsu P-K, Tsay Y-F (2013) Two phloem nitrate transporters, NRT1. 11 and NRT1. 12, are important for redistributing xylem-borne nitrate to enhance plant growth. Plant Physiol 163(2):844–856
Hu B, Wang W, Ou S, Tang J, Li H, Che R, Zhang Z, Chai X, Wang H, Wang Y (2015) Variation in NRT1. 1B contributes to nitrate-use divergence between rice subspecies. Nat Genet 47(7):834–838
Hwang E-Y, Song Q, Jia G, Specht JE, Hyten DL, Costa J, Cregan PB (2014) A genome-wide association study of seed protein and oil content in soybean. BMC Genomics 15(1):1
Johnson RC, Nelson GW, Troyer JL, Lautenberger JA, Kessing BD, Winkler CA, O’Brien SJ (2010) Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genomics 11(1):724
Jørgensen M, Bauw G, Welinder KG (2006) Molecular properties and activities of tuber proteins from starch potato cv. Kuras (journal of agricultural and food chemistry). J Agric Food Chem 54(25):9389–9397. https://doi.org/10.1021/jf0623945
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S-Y, Freimer NB, Sabatti C, Eskin E (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354. https://doi.org/10.1038/ng.548
Karn A, Gillman JD, Flint-Garcia SA (2017) Genetic analysis of teosinte alleles for kernel composition traits in maize. G3 (Bethesda) 7(4):1157–1164
Klaassen MT, Bourke PM, Maliepaard C, Trindade LM (2019) Multi-allelic QTL analysis of protein content in a bi-parental population of cultivated tetraploid potato. Euphytica 215(2):14. https://doi.org/10.1007/s10681-018-2331-z
Kloosterman B, Abelenda JA, Gomez MDMC, Oortwijn M, de Boer JM, Kowitwanich K, Horvath BM, van Eck HJ, Smaczniak C, Prat S, Visser RGF, Bachem CWB (2013) Naturally occurring allele diversity allows potato cultivation in northern latitudes. 495 (7440):246–250
Kudo K, Onodera S, Takeda Y, Benkeblia N, Shiomi N (2009) Antioxidative activities of some peptides isolated from hydrolyzed potato protein extract. J Funct Foods 1(2):170–176
Kunkel R, Campbell GS (1987) Maximum potential potato yield in the Columbia Basin, USA: model and measured values. Am Potato J 64(7):355–366. https://doi.org/10.1007/bf02853597
Léran S, Varala K, Boyer J-C, Chiurazzi M, Crawford N, Daniel-Vedele F, David L, Dickstein R, Fernandez E, Forde B (2014) A unified nomenclature of nitrate transporter 1/peptide transporter family members in plants. Trends Plant Sci 19(1):5–9
Li J, Ji L (2005) Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95(3):221–227
Ortiz-Medina E (2006) Potato tuber protein and its manipulation by chimeral disassembly using specific tissue explantation for somatic embryogenesis. PhD dissertation. McGill University. Department of Plant Science. Montreal, Quebec, Canada
Patil G, Mian R, Vuong T, Pantalone V, Song Q, Chen P, Shannon GJ, Carter TC, Nguyen HT (2017) Molecular mapping and genomics of soybean seed protein: a review and perspective for the future. Theor Appl Genet 130(10):1975–1991. https://doi.org/10.1007/s00122-017-2955-8
Pe S, Krohn RI, Hermanson G, Mallia A, Gartner F, Provenzano M, Fujimoto E, Goeke N, Olson B, Klenk D (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150(1):76–85
PGSC (2011) Genome sequence and analysis of the tuber crop potato. Nature 475(7355):189–195
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rosyara UR, De Jong WS, Douches DS, Endelman JB (2016) Software for genome-wide association studies in autopolyploids and its application to potato. Plant genome 9(2)
Rouppe van der Voort J, van der Vossen E, Bakker E, Overmars H, van Zandvoort P, Hutten R, Klein Lankhorst R, Bakker J (2000) Two additive QTLs conferring broad-spectrum resistance in potato to Globodera pallida are localized on resistance gene clusters. Theor Appl Genet 101(7):1122–1130. https://doi.org/10.1007/s001220051588
Ruseler-van Embden JGH, Van Lieshout L, Smits S, Van Kessel I, Laman J (2004) Potato tuber proteins efficiently inhibit human faecal proteolytic activity: implications for treatment of peri-anal dermatitis. Eur J Clin Investig 34(4):303–311
Sabaté J, Soret S (2014) Sustainability of plant-based diets: back to the future. Am J Clin Nutr 100(suppl_1):476S–482S. https://doi.org/10.3945/ajcn.113.071522
Segura V, Vilhjalmsson BJ, Platt A, Korte A, Seren U, Long Q, Nordborg M (2012) An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet:44. https://doi.org/10.1038/ng.2314
Sharma SK, MacKenzie K, McLean K, Dale F, Daniels S, Bryan GJ (2018) Linkage disequilibrium and evaluation of genome-wide association mapping models in Tetraploid potato. G3 (Bethesda). https://doi.org/10.1534/g3.118.200377
Tilman D, Clark M (2014) Global diets link environmental sustainability and human health. Nature 515:518–522. https://doi.org/10.1038/nature13959
Van Eck HJ, Vos PG, Valkonen JP, Uitdewilligen JG, Lensing H, de Vetten N, Visser RG (2017) Graphical genotyping as a method to map Ny (o, n) sto and Gpa5 using a reference panel of tetraploid potato cultivars. Theor Appl Genet 130(3):515–528
Voorrips RE, Gort G, Vosman B (2011) Genotype calling in tetraploid species from bi-allelic marker data using mixture models. BMC Bioinformatics 12(1):172
Vos P (2016) Development and application of a 20K SNP array in potato. PhD dissertation. Chair group: Plant breeding. Wageningen University, Wageningen, the Netherlands. Retrieved from http://edepot.wur.nl/392278. Accessed 10 Jan 2017
Vos PG, Uitdewilligen JG, Voorrips RE, Visser RG, van Eck HJ (2015) Development and analysis of a 20K SNP array for potato (Solanum tuberosum): an insight into the breeding history. Theor Appl Genet 128(12):2387–2401
Vos PG, Paulo MJ, Voorrips RE, Visser RG, van Eck HJ, van Eeuwijk FA (2017) Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theor Appl Genet 130(1):123–135
Werij JS (2011) Genetic analysis of potato tuber quality traits. PhD dissertation. Wageningen University, Wageningen, The Netherlands. Retrieved from http://edepot.wur.nl/183746. Accessed 13 Oct 2016
Willemsen JH (2018) The identification of allelic variation in potato. PhD dissertation. Chair group: Plant breeding. Wageningen University, Wageningen, the Netherlands
Acknowledgements
The authors thank the potato-breeding companies from the Centre for Biosystems and Genomics (CBSG) consortium for providing the raw data.
Funding
MTK was funded by Aeres University of Applied Sciences, Centre for Biobased Economy (CBBE), AVEBE and Averis Seeds. JHW was funded by the Dutch National Organisation for Scientific Research (NWO), under project no. 831.14.002. PGV was funded by Centre for Biosystems and Genomics (CBSG) and the breeding companies Agrico, Averis Seeds, HZPC, KWS and Meijer. These funds are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
MTK performed the GWAS, graphical genotype analysis and wrote the manuscript. JHW carried out the haplotype analysis. PGV performed the genotype calling. HJvE collected the nematode data. LMT and HJvE coordinated the project. LMT, CM and HJvE conceived the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The research described in this paper complies with the current laws of the country in which it was performed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Klaassen, M.T., Willemsen, J.H., Vos, P.G. et al. Genome-wide association analysis in tetraploid potato reveals four QTLs for protein content. Mol Breeding 39, 151 (2019). https://doi.org/10.1007/s11032-019-1070-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-1070-8