
Abstract
The discovery of nalidixic acid is one pinnacle in medicinal chemistry, which opened a new area of research that has led to the discovery of several life-saving antimicrobial agents (generally referred to as fluoroquinolones) for over decades. Although fluoroquinolones are frequently encountered in the literature, the utility of quinolone compounds extends far beyond the applications of fluoroquinolones. Quinolone-based compounds have been reported for activity against malaria, tuberculosis, fungal and helminth infections, etc. Hence, the quinolone scaffold is of great interest to several researchers in diverse disciplines. This article highlights the versatility of the quinolone pharmacophore as a therapeutic agent beyond the fluoroquinolone profile.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Throughout human history, diseases caused by microbes are often a big challenge to humanity. They are a menace to global health and the main cause of infections acquired at health care facilities—commonly referred to as hospital acquired infections (HAIs) [1]. Thus, the identification of entirely novel molecules aimed at treating such diseases is an important task [2]. Quinolone is one main antimicrobial compound class used worldwide for the treatment and management of microbial diseases [1, 3]. An attempt aimed at chloroquine synthesis in 1962, led to the accidental discovery of the first quinolone [4, 5]. The impurity was named nalidixic acid and is the predecessor of a subclass of quinolones known as the fluoroquinolones. It was later introduced in 1967 for clinical use as a treatment for urinary tract infections (UTIs) [6]. After its discovery, several generations of fluoroquinolones have been identified and developed through extensive research efforts aimed at producing compounds with improved pharmacological and drug-like properties. Today, quinolones are important class of synthetic molecules exhibiting wide spectrum of therapeutic properties [7]. Besides the numerous therapeutic applications, the quinolone nucleus can be obtained through several synthetic strategies [8]. Quinolone based compounds have been reported for activity against malaria, tuberculosis, fungal and helminth infections, etc. [9]. Hence, the quinolone scaffold is of great interest to several researchers in diverse disciplines [7]. Figure 1 below is the general quinolone core structure commonly encountered in the literature.

The development of fluoroquinolones peaked up in the 1970s and 1980s [6]. Almost all quinolone antibiotics in use are fluoroquinolones, with ciprofloxacin being the most widely used antibiotic worldwide. They are so called because of the fluorine at C-6 position of the quinolone core, which was one of the earliest changes to the structure of nalidixic acid. Fluoroquinolones have a wide spectrum of activity against Gram-negative and -positive bacteria [5]. Fluoroquinolones are categorised into first, second, third, and forth generations, (see Fig. 2 below), with more than10,000 analogues synthesised since the discovery of nalidixic acid [11]. In this review, fluoroquinolones will be overlooked, and focus shifted to recently discovered subclasses of quinolones with the aim of highlighting the therapeutic versatility of quinolone scaffold in different diseases and highlight on the different synthetic protocols reported for the synthesis of this scaffold.

Generations of fluoroquinolone-based drugs
Quinolone synthesis
The quinolone core is present in several therapeutic compounds. Hence, many researchers are interested in the synthetic procedures that afford novel structurally diversified quinolone derivatives [12]. Highlighted below are few examples of synthetic procedures commonly deployed in the synthesis of quinolone containing compounds.
After George Lesher and co-workers accidentally discovered nalidixic acid, they published an article on the investigation of nalidixic acid derivatives in 1962. The reported synthetic procedure involves reacting 6-substituted-2-aminopyridine (13) and diethyl ethoxymethylenemalonate to form a condensed intermediate (14), which is subsequently cyclized using Dowtherm A or diethyl phthalate to give the ethyl 4-hydroxy-1,8-naphthridine-3-carboxylate derivative (15). Consecutive Ester hydrolysis and N-alkylation afford the desired 1-alkyl-1,8-naphthyridin-4-one-3-carboxylic acid (16) [4] (see Scheme 1).

Synthesis of 1,8-naphtharidine (nalidixic acid derivatives) derivatives
Shi et al. reported the use of Co(III) catalyst for the synthesis of quinolone derivatives. Briefly, experimental conditions for reacting (E)-3-dimethylamino-phenylprop-2-en-1-one (17) and 3-phenyl-1,4,2-dioxazol-5-one were optimized. The yield was low with [Cp*Co(Co)I2], AgSbF6 and KOAc, AgOAc, or HOAc in dichloroethane (DCE) after 12 h. No reaction occurred in the absence of [Cp*Co(Co)I2] and AgSbF6, suggesting that Co(III) is an essential catalyst for this synthesis. Reaction yields increased when AgSbF6 was used as an additive. Moreover, reaction yields were highest when DCE was used as solvent and AgBF4 as salt. The advantages of this method include fewer reaction steps and short reaction time. Moreover, the method makes feasible the generation of structural diverse quinolones (18) and it is compatible with various functional groups [13] (see Scheme 2).

Co (III)-catalysed synthesis of quinolones [13]. Reaction conditions: 17 (0.2 mmol) Cp*Co(CO)I2 (10 mol%), AgBF4 (20 mol%), DCE (2 ml), 80 °C, 12 h
Considerable attention has recently been given to microwave-assisted synthesis of various organic compounds as this method seems to afford target compounds in high yield, shorter reaction times and high purity [14]. One example is the synthesis of 2-methyl-4-quinolones with varied substituents on the benzenoid ring reported by Duarte et al. Target compounds were achieved through microwave irradiation of different anilines (19) and ethyl acetoacetate (20) using diphenyl ether as a solvent at 205–230 °C for 5 min at 300 W in a one-step (see Scheme 3) to afford target compounds (21). The key features for the synthesis were high yields, reduced reaction time, and easily accessible starting material [15].

Microwave assisted synthesis of 2-methyl-4-quinolones
Zhao et al. reported the use of palladium as a catalyst in the tandem amination protocol for the direct conversion of various o-haloaryl acetylenic ketones (22) into N-aryl-4-quinolones (23). Pd2(dba)3 was used as a catalyst, PPh3 as a ligand, dioxane as a solvent in the presence of K2CO3 and the yields were generally good [16] (see Scheme 4).

Palladium catalysed synthesis of quinolones [16]. All reactions were performed under N2 on a 0.5 mmol scale, using aryl amines (1.2 eq), Pd2(dba)3-CHCl3 (5 mmol%), PPh3 (10 mol %), and K2CO3 (2.0 eq) in dioxane (3 mL) under reflux. The isolated yields were up to 93%
Shao et al. reported the synthesis of N-alkyl-substituted-4-quinolones (24) in high yields via palladium-catalysis using an alternative method that makes use of o-chloroaryl acetylenic ketones and alkyl amines. Among the tested bases, K3PO4 proved to be the best with DMSO as a solvent at 1400C. Quinolones bearing diverse functional groups were obtained using this method [17].
Xu et al. reported the synthesis of 4-quinolones catalysed by Cu(OTF)2. Reagents employed in this study included secondary anilines (25) and alkynes (26). The best conditions for this method are 5 mol % Cu (OTF)2, 5 mol % HOTf (trifilic acid), dichloromethane (DCE), temperature of 1200C and a reaction time 12 h. Xu and co-workers observed that 4-quinolones (27 and 28) in excellent yields were obtained using N-alkyl and N-aryl substituted anilines. The key features for this reaction include the use of mild reaction conditions, high functional group tolerance and its applicability for scale-up [18] (see Scheme 5).

Cu-mediated synthesis of 4-quinolones [18]. Reaction conditions: 25 (05 mmol), 26 (0.6 mmol), Cu(OTF)2 (0.025 mmol), HOTF (0.025 mmol), DCE (2 mL), 120 °C, 12 h
Akerbladh et al. reported the synthesis of 4-quinolones from 2-iodoanilines and alkynes through palladium catalysis. This procedure does not require CO and it involves carbonylative, Sonogashira coupling/cyclization sequences. They deployed two methods to achieve target compounds. In the first method, phenylacetylene (29), 2-iodoaniline (30), Mo (CO)6, and diethylamine in a seal vial were irradiated in a microwave at 120 °C for 20 min. The yields were 76% when Pd(dppf)Cl2 was used as a catalyst and sodium acetate as a base. When Pd2(dba)3 and an excess of dppf were used, the yield increased to 85%. The second method was performed at room temperature and hence suitable for heat sensitive molecules. For both methods, CO was generated in situ from molybdenum hexacarbonyl catalysed by palladium [19] (see Scheme 6).

Palladium-catalysed synthesis of 4-quinolones [19]. Reaction conditions: 2-Iodoaniline (0.5 mmol), phenylacetylene (1 mmol), base (1.5 mmol), MO9CO)6 (1 mmol), Et2NH, 120 °C, 20 min, 10 mol % of palladium catalyst. 12–13 entries up to 85% yields
Hu et al. reported on metal-free synthesis of 2-arylquinolones (34) using readily available N-arylmethyl-2-aminophenylketones (33). The best conditions affording up to 98% yields include the use of TEMPO as the oxidant and DMSO as a solvent with KOtBu as a base at 800C for 6 h. Advantages of this method includes no use of transition metal catalyst, mild conditions, and high yields [20] (see Scheme 7).

TEMPO catalyst synthesis of 2-arylquinolones [20]. Reaction conditions: 33 (0.5 mmol), KOtBu (1 mmol), TEMPO (1 mmol), DMSO (10 mL), 80 °C, 6 h. 20 examples with up to 98% yields
Kang and Hong reported the synthesis of C5 or C2 alkynylated 4-quinolones (35 and 36). Briefly, they used an alkylating agent, TIPS-EBX, to execute C5 selective alkynylation of 4-quinolones. C2 selective alkynylation was achieved using N-pyrimidyl group-directed cross coupling [21] (see Scheme 8).

Site selective alkynylation of quinolones [21]. Reaction conditions: quinolone (35. 0.1 mol), Tips-EBX (1.1 eq), [RhCp*(MeCN)3(SbF6)2] (5 mol %) in toluene at 8 °C for 12 h under air, yields up to 81%
Satio et al. reported the synthesis of 2-substituted dihydro-4-quinolones in high yields using 2-aminophynyl vinyl ketones (38) as substrates and chiral phosphoric acid as catalyst. The product is formed through consecutive aza-Michael addition and oxidation. Enantioenriched azaheterocycles are often obtained through aza-Michael addition, however, this reaction faces the challenge of generating compounds with high stereo-selectivity. This challenge is often overcome using stereo-directing groups, which need to be removed at the end of the synthetic process—this thus increases the synthetic steps and cost. As a result the development of stereo-directing catalyst in aza-Michael addition is highly desired to decrease the number of reaction steps [22] (see Scheme 9).

Chiral phosphoric acid catalysed synthesis of 2-substituteddihydro-4-quinolones [22]. Reactions were carried out on a 0.1 mmol scale with starting material (0.1 mmol) and (R)—3 g (10 mol%) in benzene (0.5 mL)/cyclohexane (0.5 mL) at 70 °C, quantitative yields up to 94%
Monastyrskyi et al. reported the synthesis of quinolones from ethyl acetoacetate (EAA) (41) mediated by hypervalent diaryliodonium salts. Their method requires mild conditions, readily available reagents like EAA, and does not require the use of metal catalyst. The method requires treating iodonium salts and EAA in DMF with a base such as tBuOK, or Cs2CO3. The resulted compounds (42), which is a 2-aryl EAA, converted to the corresponding 3-aryl-4(1H)-quinolones (43). This method can be viewed as modified Conrad-Limpach cyclization. This method was recently used to synthesis ELQ300, a clinical candidate for the treatment of malaria [23] (see Scheme 10).

Synthesis of quinolones mediated by diaryliodonium salt [23]. Reaction conditions: salt was added to the enolate solution of EAA and ran for 18–24 h, yields were up to 60%
Wang et al. reported a mild N–H activation/Heck reaction wherein benzyne precursors (44) and acrylamides (45) were conveniently transformed to a variety of quinolones (47) in a one step procedure catalyst by palladium. Several reaction conditions were sampled and the best reaction conditions identified is; 5 mol % of Pd(OAc)2, Cu(OAc)2 (2 eqv), C5F (2.4 eqv), dioxane and DMSO solvent mixture, a temperature of 80 °C and a reaction time of for 24 h [24] (see Scheme 11).

Palladium-catalysed synthesis of quinolones from acrylamides and benzene precursors [24]. Reaction conditions: 44 (0.4 mmol), 45 (0.2 mmol), Pd(OAc)2 (5 mol%), Cu (OAc)2 (0.4 mmol), CsF (0.48 mmol), TBAB (0.2 mmol), molecular sieves (100 mg), DMSO/dioxane mixed solvent at 80 °C for 24 h. Pd(OAc)2 (10 mol%); the solution of 45 in 1.5 mL solvent was added slowly via syringe pump in 15 h
Manikanda and Jeganmohan reported the synthesis of diverse 4-substituted-2-quinolones from anilides (48) and propiolates or acrylates using Ruthenium-catalyst. The target quinolones were achieved in good to excellent yields. They further effected halo group (Cl or Br) addition at position C3 of the quinolones (50) using either NBS or NCS as the halogen source. In addition, the 2-quinolones were converted to 2-chloroquinolines using POCl3 as the chlorinating agent. They also pointed out that indole and pyrrole derivatives could be obtained by treating anilines or enamines with alkynes in the presence of a rhodium or ruthenium as catalyst [25] (see Scheme 12).

Synthesis of 2-quinolones from anilides catalysed by Ruthenium [25]. All reactions were carried out using 48 (100 mg), ethyl-2-butynoate (1.5 eq), [[26]2] (0.05 eq), AgSbF6 (0.20 eq), and Cu (OAc)2, H2O (1.5 eq) in DCE 2.5 mL at 110 °C for 12 h. After that, 30% of HCl was added and heated at 130 °C for 10 h, yields up to 83%. The first step was only allowed for 5 h after that, 30% of HCl was added and heated at 130 °C for 5 h
Therapeutic applications of quinolone containing compounds
Quinolones showing activity against malaria parasite and related apicomplexan parasites
Malaria, a life-threating disease, is caused by infection with Plasmodium parasites. These parasites are typically transmitted when an infected Anopheles mosquito bites a human for a blood meal. In humans, the Plasmodium parasite first site of growth and multiplication is the liver cells, from where they then enter red blood cells and multiply exponentially. Malaria symptoms appear during this exponential growth phase. Eleven (11) countries, mostly in Africa, accounts for approximately 70% of the world’s malaria burden and children younger than 5 years account for 61% of malaria deaths worldwide—making them the most vulnerable group [27]. Although Malaria is preventable and treatable, artemisinin (the cornerstone of anti-malarial treatment) resistance has emerged and this puts malaria control and elimination efforts at jeopardy [28, 29]. Thus, novel compounds inhibiting new parasite targets are urgently required to sustain effective treatments against malaria. Quinolone based compounds have been reported to demonstrate potent activity against malarial parasite and hence are potential antimalarial agents [30]. They show good efficacy and exhibit mul- targets activities against malarial parasite. Quinolone derivatives are active against the blood, liver and gametocyte stages of malarial parasites [31].
Endochin (53), a 4-(1H)-quinolone, was initially investigated as an antimalarial drug against avian malaria affecting canaries, wherein P. cathemerium is the parasite. It was described seventy years ago. Endochin inhibits parasites’ cytochrome bc1 complex and this consequently alters the normal functioning of the parasite’s mitochondrial electron transport chain (ETC), a validated target for atovaquone [32]. The high activity of endochin against avian malaria model gave hope to the discovery of an entirely novel class of antimalarial agents. Unfortunately, endochin and its derivatives were not effective when tested against malaria in humans, and research interest in them was discontinued. Then in the early 2000s, this compound class was re-examined, and a new class of compounds called Endochin-like quinolone (ELQ) were discovered. ELQs are reported to be very potent against apicomplexan pathogens in vitro. They are also efficacious in animal models of acute and latent apicomplexan infections, including Babesia, Toxoplasma gondii, and Plasmodium falciparum [33]. ELQs have a unique chemical space around the quinolone nucleus, which is characterized by the presence of a methyl group and a diphenyl ether substituent at position 2 and 3, respectively. This is in addition to other substituents in the benzenoid ring [34]. ELQ-300 (54) and its prodrugs (ELQ-331 and 337) are reported to be active against the liver and blood stages of P. falciparum and, also against gametocytes, zygotes, and ookinetics. These stages are crucial for disease transmission [35]. ELQ-331 (55) is currently a preclinical candidate that is being developed by Medicines for Malaria Venture. It is being developed as a potential agent for malaria prevention and treatment. In addition, ELQ-316 (56) and its prodrugs displayed potent activities versus T. gondii and Babesia microti [34]. As reported by Nilsen et al., a single oral dose (0.03 mg/kg) of ELQ-300 prevented infections in malaria mouse models and four daily doses of 1 mg/kg resulted in complete cure of infected mouse models of malaria. These results indicate that ELQ-300 has acceptable pharmacokinetic properties that allow complete cure of mice when given orally and is effective in blocking transmission in rodent models of malaria [35].
Although ELQ-300 shows potent antimalarial activity in vitro and in vivo, it has some poor physicochemical properties. For example, its relatively high crystallinity limit aqueous solubility and absorption. Consequently, very low blood concentrations are obtained after oral dosing. Thus, developers have turn to the prodrugs strategy to resolve the challenges associated with its poor physicochemical properties. Milley et al., reported the pharmacological profiling of ELQ-337 (57), a bio-reversable, prodrug of ELQ-300. In comparison to ELQ-300, with molar equivalent dose of 3 mg/kg body weight, the prodrug led to three-to fourfold increase in bioavailability following oral administration, reaching a maximum concentration of drug in serum (Cmax) of 5.9 µM. In addition, a single low dose (in milligrams) of the prodrug cured patent malaria infections in mice. These results suggest that prodrug approach may overcome the physicochemical liabilities associated with ELQ-300 [36]. In another study by Frueh et al., aimed at overcoming the challenges associated with ELQ-300’s poor physiochemical properties, they synthesised several bio-reversible ester prodrugs of ELQ-300. These agents were converted to ELQ-300 by esterase, which are abundant in the liver and bloodstream of the host. One of these, ELQ-331, at a single dose of 3 mg/kg cured patent malaria infection in a murine model infected with P. yoelli. Moreover, the result showed that ELQ-331 is a ELQ-300 prodrug with improved physiochemical and metabolic properties suitable for clinical studies [37].
Dogget et al., identified ELQs-316 as a lead compound for toxoplasmosis. They noted that this compound showed specificity for T. gondii cytochrome b over human cytochrome b. In this study, a carbonate ester prodrug of ELQ-316, ELQ-334 (58), was synthesised with the aim of overcoming the poor pharmacokinetic properties of ELQ-316. Oral administration of ELQ-334 resulted in a sixfold increase in both the maximum plasma concentration (Cmax) and the area under the curve (AUC) of ELQ-316. The increased bioavailability of ELQ-316, when administered as ELQ-334, ultimately led to efficacy against latent toxoplasmosis which is comparable to ELQ-316 administer intraperitoneally. These findings suggest that carbonate ester prodrugs could be a useful strategy to overcome the poor pharmacokinetic properties of ELQs for the treatment of toxoplasmosis [33]. Toxoplasmosis is a common parasitic infection affecting humans in developed countries, and it is a fatal infection to developing foetus and people with compromised immune systems [38, 39]. Its prevalence varies between 20% and 80%, and is influenced by age, cultural practices, and environmental factors [40].
In a study reported by Eberhard et al., the activity of different ELQs against B. besnoiti tachyzoites grown in human foreskin fibroblasts were established through quantitative real time polymerase chain reaction (PCR). The result showed that the ELQs under evaluation exhibited lower activity against B. besnoiti compared to activities observed against P. falciparum and T. gondii. However, the B. besnoiti and T. gondii share high sequence similarity for their cytochrome b. Moreover, the predicted ELQ binding sites (Qo and Qi) within the cytochrome b of toxoplasma, neospora, and Besnoiti are virtually identical. These suggest that the variation in ELQ activities against these pathogens is not target related.
Complete cure of B. basnoiti infection with ELQs seems to require longer treatment period and/or larger doses of ELQs. For example, it has been observed that after 20-days of being exposed to ELQs, B. basnoiti parasites resume proliferation a few days after exposure to ELQs was stopped [41] (see Fig. 3).

Endochin and endochin-like quinolones with activities against apicomplexan parasites
ICI 56780 (59), another 4(1H)-quinolone lead against malaria, was discovered by Ryley and Peters in 1970. This compound was reported to exhibit activity against multiple targets across different stages of P. falciparum parasite. However, this compound was abandoned because it suffered from poor aqueous solubility and it precipitated the development of resistant P. berghei parasite in mice treated just once with the compound [42]. Later, ICI 56780 was observed to have anti-relapse activity in vivo and it also demonstrated excellent blood stage activity. These data ignited renewed interest in this compound and researchers once again became interested in its multistage antimalarial activity. Cross et al., used a parallel approach of SAR and pharmacological characterization to design IC 56780 analogues void of cross-resistance with atovaquone (60). They generated several quinolones with potent antimalarial activity, with some of the compounds having IC50 values as low as 0.15 nM against P. falciparum [43].
Using SAR and structure property relationship (SPR), Neelarapu et al., conceptualized a synthetic strategy to incorporate a piperazinyl moiety onto the quinolone scaffold. The lead compound from this study (61) had good antimalarial activity, aqueous solubility, and oral bioavailability. The lead compound was evaluated for in vivo efficacy aimed at the blood stages of the parasite. The results showed an in vitro EC50 of 5 nM versus P. berghei, with in vivo % inhibition (100 mg/kg) > 99% curative in 5 mice for 30 days [44].
Cowley et al., reported the synthesis of novel 6-and 7- substituted quinolone (62) via the Gould-Jacobs methodology. The Qo site in cytpchrome bc1 complex, where ubiquinol is oxidised was the site of interest in this study. The target compounds generally contain a polar ‘head’ group and an aryl ‘tail’ at position 3 and 6/7 of the quinolone nucleus, respectively. In this study, novel quinolones compounds bearing aryl ‘tail’ at position 7 of the quinolone nucleus showed potent antimalarial activity as low as 0.46 nM against P. falciparum. Molecular docking studies demonstrated that molecules forming strong interactions with residues His 182 and Glu 272 had high potency [45].
Decoquinate (63), a quinolone currently used as a coccidiostat in poultry, has a single digit nanomolar activity against blood stages of P. falciparum parasite in in vitro assays. In a study by Nam et al. wherein known drugs, and other compounds were screened; decoquinate was identified as an antimalarial agent with high therapeutic index. These inspired further studies aimed at elucidating the molecular target of decoquinate. Target discovery studies pinpointed that this compound targets the ubiquinol-binding pocket of P. falciparum cytochrome b [46]. Decoquinate’s poor solubility in water and/or any other organic solvent prompted a group of chemist to convert this drug into new tractable derivatives, some of which demonstrated good in vitro antimalarial activity against chloroquine-sensitive (NF54) and multidrug-resistant (K1 and W2) P. falciparum strains. The best compound from this study, the N-acetyl derivative (64), was 50-fold more active than decoquinate; exhibiting IC50 values in the range of 0.32–1.5 nM against apicomplexan parasites [47, 48].
Suet et al. reported SL-2-25 (65), an antimalarial lead compound whose mode of action involves inhibition of NADH: ubiquinone oxidoreductase (PfNDH2) of P. falciparum. In this study, they attached several heterocycles to the quinolone core in a bit to generate CK-2-68 analogues with reduced ClogP and improved aqueous solubility. SL-2-25 emerged as the lead compound, having IC50 values in the nanomolar range versus both the PfNDH2 enzyme and whole cell P. falciparum (IC50 = 15 nM PfNDH2; IC50 = 54 nM 3D7 strain of P. falciparum). Its phosphate salt was reported to exhibit notable oral activity of ED50/ED90 of 1.87/4.72 mg/Kg versus P. berghei (NS strains) in a murine model of malaria. Moreover, compounds in this study also inhibited the bc1 complex of P. falciparum with nanomolar activity; and as such provide dual mechanism of action against malaria parasite. The potent oral activity of 2-pyridyl quinolones underlines the potential of this template for further lead optimization [49] (see Fig. 4).

Quinolones showing activity against malaria parasite and related apicomplexan parasite
Quinolones on TB and HIV
Mycobacterium tuberculosis (Mtb), the causal agent of tuberculosis (TB) is reported to currently infect approximately one third of the world’s population [50]. Although TB is a prehistoric disease, it continues to pose as a global public health threat. TB is currently among the top 10 causes of death and the second leading cause of death from a single pathogenic agent, second only to COVID-19 [51, 52]. The greatest impact on TB associated fatalities was projected in 2021 wherein the number of deaths is on par with 2017 records, while the greatest impact on tuberculosis incidence is expected in 2022 [52]. Although nonadherence to first line TB treatment poses a serious threat to TB control, the upsurge in TB prevalence is fuelled by the acquired immune defiance syndrome (AIDS) epidemic. AIDS, caused by two distinct human immunodeficiency viruses (HIV), HIV type1 and 2 (HIV-1/HIV-2), is one of the largest and most devastating public health pandemics. While HIV-1 is responsible for the great majority of infections globally, HIV-2 infection is very rare outside of West Africa [53]. HIV suppresses the immune system and increases the risk of opportunistic infections, including TB [54]. The gradual loss of CD4+ T cells from blood, lymphoid organs, and mucosal tissues is a hallmark of untreated HIV infection [55]. The common routes of HIV transmission includes, sexual transmission (anal, oral and vagina sex), mother to child transmission, sharing of contaminated needles [53]. The current World Health Organization (WHO) estimate suggests that 36.9 million people (35.1 million adults and 1.8million children) worldwide are living with HIV, with more than 1.5 million new cases and around 1 million deaths reported annually [56]. Studies have shown that HIV infection precipitates the development of active TB [57]. For example, nations which previously had TB under control are witnessing dramatic increases in TB rates as HIV infection rates rises. Following this observation, experts have warned that tuberculosis cannot be controlled using existing tools which do not take HIV-coinfection into consideration [50]; hence the quest for new treatment options for these diseases still persist. Herein we present quinolone-based compounds reported for activity against Mtb or HIV.
Quinolones active against the causal agent of TB
The antibacterial activity of quinolones versus a variety of Gram-negative/positive bacteria has been reported by many researchers [58, 59]. Fluoroquinolones (Fig. 2 above) have in vitro activity against Mtb with the newer quinolones (sparfloxacin, gatifloxacin, and moxifloxacin) having lower minimum inhibitory concentration (MIC) compared to levofloxacin, ciprofloxacin, and ofloxacin [60]. Moreover, the WHO approved fluoroquinolones for the treatment of multidrug-resistant TB (MDR-TB) because they have potent and broad-spectrum activity. Fluoroquinolones suppress Mtb growth by inhibiting it’s DNA gyrase and topoisomerase IV, as is the case in other bacteria [61]. Furthermore, they have excellent pharmacokinetic properties and have been reported to penetration into host macrophages [3]. The global coverage of fluoroquinolone resistance testing remains significantly low, a little over 50% in 2020 [52]. Nijland et al. reported that rifampicin reduces plasma concentration of moxifloxacin in TB patients. They investigated the interaction between rifampicin and moxifloxacin and observed that coadministration of moxifloxacin with rifampicin and isoniazid resulted in reduced plasma concentration of moxifloxacin. Thus, they concluded that there exist drug-drug interaction following coadministration of moxifloxacin and rifampicin, which negatively affects the efficacy of moxifloxacin [62].
Besides fluoroquinolones, other quinolone subclasses with activity against Mtb are increasingly being reported, with some exhibiting a unique mode of action, different from that reported for the fluoroquinolone subclass.
Senthilkumar et al. identified novel 6-nitroquinolones bearing carboxylic acid moiety at position 3 of the quinolone nucleus. Several nitroquinolone-3-carboxylic acids were synthesised through a six-step synthesis and evaluated in vivo and in vitro for antimycobacterial activity against drug susceptible and drug-resistant strains of Mtb and M. smegmatis. These compounds were also evaluated against DNA gyrase from Mtb smegmatis. Compound 66 emerged as the lead compound, exhibiting MIC values of 0.08 and 0.16 µM against Mtb H37Rv and MDR-TB, respectively. The decrease in MIC against MDR-TB suggests this compound mode of action is like that of fluoroquinolones. Furthermore, at the dose of 50 mg/Kg body weight, the lead compound decreased bacterial load in lung and spleen tissues with 2.78 and 4.15-log protections, respectively [63].
Beteck et al., reported the anti-tubercular activity of 6-nitroquinolone bearing carboxylate ester at position 6 of the quinolone nucleus. Mutant enzyme inhibition and molecular docking studies showed the hit compound from this study as DprE1 inhibitor. However, the compounds were screened as suspensions because they were poorly soluble in screening media. Thus, no structure–activity relationship (SAR) analysis could be done for this study [64]. In continuation of this work, Dube et al., generated 6-nitroquinolone-3-carboxamides derivatives; attaching polar moieties of different chain lengths and ring sizes at position C3 of previously insoluble compounds. All target compounds were evaluated in vitro for anti-TB activity using the gfp reporter strain of Mtb and twelve compounds exhibiting antitubercular activity in the range of < 0.244–31.865 µM were identified. The hit compound (67) had a MIC90 of < 0.244 µM and docking studies showed that this compound binds to DprE1 active site, with its nitro group in proximity with Cys 387 residue [51].
Decaprenylphosphoryl-β-D-ribose 2ʹ-epimerase (DprE1) is a critical enzyme in the synthesis of arabinogalactan, which is an essential molecule needed for the formation of mycobacteria cell wall [65]. Thus, DprE1 is a promising drug target for exploring various chemotypes with cidal activity against Mtb. In addition, there are currently no marketed DprE1 inhibitors in use for the treatment of TB. Thus, compounds targeting DprE1 do not have the challenge of overcoming pre-existing drug resistance; making DprE1 an attractive target for developing effective and safer medicines for the treatment of all strains of Mtb. Furthermore, DprE1 is specific to mycobacteria and actinomycetes and is absent humans, reducing the risk of unwanted side effects [66]. Naik et al., reported 4-aminoquinolone piperidine amides (AQs) (68) as a novel scaffold, identified from a whole cell screen, with potent inhibition on replicating as well as slow growing Mtb. Evaluation of the minimum inhibitory concentrations, followed by whole genome sequencing of mutants raised against AQs identified DprE1 as the primary target responsible for the antitubercular activity. Mass spectrometry and enzyme kinetic studies indicated that AQs are noncovalent, reversible inhibitors of DprE1 with slow on rates and long residence times of ~ 100 min on the enzyme. Generally, AQs show outstanding lead-like characteristics and a favourable secondary pharmacology profile in vitro. Although the scaffold started off as a single active compound with moderate potency from the whole cell screen, structure—activity relationship optimization of the scaffold led to compounds with potent DprE1 inhibition (IC50 < 10 nM) along with potent cellular activity (MIC = 60 nM) against Mtb [66].
Beteck et al. reported on decoquinate (DQ) derivatives showing activity against Mtb (69). They derivatised DQ through trans esterification or aminolysis of the ethyl ester at position 3 of DQ to generated derivatives with more polar alkoxy esters or less readily metabolized amides. Polar groups were also attached to the quinolone nucleus at N-1 or O-4. DQ is a coccidiosis therapy used in veterinary medicine that has a favourable safety profile. It is inexpensive but has a high lipophilicity and a very low aqueous solubility (0.06 g/mL). To generate SARs around the benzenoid ring of DQ, they prepared shorter chains as opposed to the dodecyl chain in DQ. All derivatives and shorter chain analogues were evaluated in vitro against Mtb. The N-alkyl quinolone amides exhibited potent activity against Mtb with MIC90 values ranging from 1.4 to 3.64 µM, whereas DQ was inactive [47]. Disruption of cell wall homoeostasis is proposed as one possible mode action for these compounds. These derivatives are not active against lung fibroblast cells (With IC50 values of 40 to > 100 μM), and as such have selective activity toward Mtb. Short chain analogues were inactive against Mtb [47].
Mtb requires adenosine triphosphate (ATP) to mediate several physiological functions, persistence, and pathogenicity. There has been a recent resurgence of interest in the electron transport chain (ETC) and adenosine triphosphate (ATP) synthesis (oxidative phosphorylation) as new target spaces for drug development [67]. Energy generation processes in mycobacteria, particularly those involved in oxidative phosphorylation pathway are currently being pursued as novel drug targets. Drug targets in energy generation have been reported to be vital to both replicating and dormant Mtb, thus appropriate lead for these targets can shorten and simplify TB chemotherapy. One such target is Adenosine triphosphate synthase, a validated target for bedaquiline [68].
By leveraging on lead repurposing strategy, Chong et al., investigated SCR0911(70) as potential mycobacterial cyt-bcc complex inhibitor. They further demonstrated that this compound has activity against fast- and slow-growing M. smegmatis and M. bovis, respectively. This lead was not active against cytochrome bcc deficient mutant strain, validating the cytochrome bcc oxidase as the target [67].
Research has shown that targeting Mtb’s respiratory chain components is effective in sterilizing Mtb. Targeting Mtb’s respiratory-chain provides a novel avenue in Mtb chemotherapy which is completely different from how standard antitubercular agents work, most of which preferentially target Mtb’s replication machinery. NDH, a 50 kDa enzyme involved in ATP synthesis, is a promising drug target against Mtb. Nicotinamide adenine dinucleotide dehydrogenase (NDH) has been discovered to be essential for cell function and viability using biochemical assays. NDH is an important and a viable drug target since Mtb cannot survive mutations at ndh gene [69] (see Fig. 5).

Quinolones with activity against Mtb
Hong et al. reported the identification of heterocyclic quinolones targeting the respiratory chain of Mtb. They screened several compounds against NADH:menaquinone oxidoreductase (NDH) of Mtb through high-throughput screen (HTS). Based on the known phenothiazine NDH inhibitors, trifluoperazine and thioridazine, 11,000 molecules were chosen for the HTS. About 100 hits from four distinct chemotypes were identified, with the quinolone core being the most promising. Following this observation, 350 more quinolone-based compounds were screened, leading to the identification of 90 more hit compounds. Quinolone hits bearing a cyclic amine moiety at position 2 were chosen as the template for hit to lead optimization studies, which culminated in the identification of the lead compound, (71). This compound displayed potent antitubercular activity (Mtb IC50 = 525 nM, Wayne Mtb IC50 = 76 nM, and MDR Mtb patient isolates IC50 = 140 nM) and has poor to modest pharmacokinetic properties [69].
Quinolones active against HIV
The HIV-1 capsid has for some time now been the target against which new anti-HIV agents are development. It is important in the maturation and assembly of HIV-1. A considerable amount of quinolone derivatives has been screened in the last three decade for activity against HIV, some of which exhibited promising potency [54]. Elvitegravir (72) is a quinolone-3-carboxylic acid and one of the first integrase (IN) inhibitors to be approved for the treatment of HIV infection by the U.S food and drug administration [70]. IN inhibitors are a class of antiretroviral (ART) that inhibits the viral IN. This viral enzyme (IN) integrates viral genome (in double-stranded DNA) into human genome; this is important for HIV replication [71]. Hence, elvitegravir inhibits an important process in HIV. It has potent activity (IC50 of < 0.06 µM/mL in vitro) [71].
In 2009, Shimura et al. reported elvitegravir and its derivatives as emerging IN inhibitors, having potent antiviral activity and favourable pharmacokinetic properties [72]. To expand on the SAR of hydroxylquinolone-3-carboxylic acid (HQCAs), He et al., synthesised and reported 5-hydroxylquinolone-3-carboxylic acids bearing varied aryl substituents on N-1 position. These compounds displayed low micromolar activity when evaluated for antiviral activity against wild-type HIV-1 (LAI strain IIIB). Compound 73a was active against wild-type HIV-1 and HIV-1 mutant (A17), exhibiting EC50 values of 3.17 and 17.88 µM, respectively. Docking studies suggested that the physicochemical properties and antiviral potency of HQCAs are influenced by the nature of N-1 substituents [73]. Notably, previous chemical modifications of the quinolone core to generate antiviral agents were made on N-1, C-6,7, and 8 positions, with very few modifications focus on the C-5 position. Therefore, He and co-workers became interested at the C-5 substituted quinolone derivatives and ultimately identified 5-hydroxylquinolone-3-carboxylic acids (73b), having low micromolar antiviral activity against HIV-1 [74].
Curreli et al., reported diverse classes of inhibitors that efficiently inhibited the formation of mature viral particles—verified using electron microscope (EM). They aimed at compounds that target the hydrophobic cavity of the c-terminal domain (CTD), a validated target for peptide-based inhibitors. The obtained results demonstrated that these compounds prevent virion maturation. Compound 74 exhibited high selectivity index (SI) of > 60 (SI = CC50/IC50) and showed good antiviral activity in the range of 1.06–12.4 µM [75].
Costi et al., identified compound 75, a quinolinyl diketo acid derivatives as HIV-1 IN inhibitors (75). Diketo acid (DKA) derivatives bearing a p-fluoro benzyl group on quinolone nitrogen and a variable base-like functional group at position 7 of the quinolone ring were synthesised and biologically tested, leading to the identification of three compounds with IC50 values below 100 µM against HIV-1 IN [76].
Kashiwase et al., investigated several fluoroquinolones for inhibitory activity against HIV-1 replication and noted that compound 76 (R-71762) reduced HIV-1 multiplication in infected cells and protected infected MT-4 cells from viral cytopathic effects [77]. Moreover, a study by Hagihara et al. identified R-71762 to have low micromolar antiviral activity against HIV-1 with IC50 of 1.7 µM. The compound’s anti-HIV activity was also tested against HIV-1IIIB chronically infected MOH-4-cells [78].
Yoneda et al., reported oxoquinoline-acylhydrizone derivatives as potential HSV-1 polymerase inhibitors. They used molecular modelling to guide the synthesis of 1,4-dihydro-4-oxoquinoline derivatives containing ribonucleosides active against both HSV-1 and HIV-1. The 6-Cl substituted derivative (77) exhibited high selective index. The mechanism of action for this agent includes binding to HSV-1 POL complex [79].
Jadulco et al., isolated three quinolone alkaloids from Melochia odorata and screened them in vitro for anti-HIV activity. At concentrations of 56.2 and 0.84 µM, Waltheriones A (78a) and C (78b) exhibited cytoprotective activities against HIV. These compounds also inhibited HIV P24 formation at less than 2.0 µM concentration [80].
Massari et al. identified 6-des-fluoroquinolones (6-DFQs) as a promising class of compound for the control of the latent HIV-1 reservoir (79). 6-DFQs exemplified by 79 displayed very good antiviral activity against HIV-1 in various human cells [81]. Following the above study, Cecchetti et al., also evaluated a series of 6-amino quinolone compounds against HIV. The most active compound (80) inhibited HIV replication in infected C8166 human lymphoplastoid cell lines, exhibiting an EC50 value of 0.1 µM [82]. Parolin et al., investigated the antiviral mode of action of 80 and found that the reverse transcriptase, integrase, and protease activities were not impaired by 6-aminiquinolones. The aminoquinolone complex TAR RNA at low nanomolar dissociation constant (19 ± 0.6 nM). These results suggest 6-aminiquinolones as new class of antiviral compound targeting viral RNA [83] (see Fig. 6).

Anti-HIV quinolones
Quinolones active against the causal agents of sleeping sickness (Human African trypanosomiasis), Chagas diseases, and leishmaniasis
Trypanosomatid illnesses exhibit a wide range of epidemiological and clinical patterns. Human Tryponosomiasis is solely found on the African continent, while Chagas disease and Leishmaniasis are widespread. The occurrence of these diseases is intimately linked to environmental and socioeconomic factors. Furthermore, the three diseases are associated with poverty [84]. Human African Trypanosomiasis (HAT), together with Chagas disease (CD), and Visceral leishmaniasis (VL), are caused by unicellular parasitic protozoa of the genus Trypanosoma, and Leishmania, respectively [85]. According to WHO, these three diseases are a threat to the health of millions of people and are responsible for more than 30,000 annual deaths; however, little research investment is made toward these diseases, thus WHO have classified them among neglected tropical diseases [85].
Human African trypanosomiasis
HAT also known as a sleeping sickness is a parasitic disease transmitted by tsetse flies. It is a serious life-threatening disease occurring in 36 African countries. T.b. gambiense is widespread in 24 of these nations and it causes West African sleeping sickness, whereas T.b. rhodesiense (a related sub species) is present in 13 countries and it causes the more severe East African sleeping sickness syndrome, with Uganda being the only country reporting both forms of the disease. If there is no pharmacological intervention, these parasites cross the blood–brain barrier (BBB), inevitably leading to sleep disorder and death [86].
Chagas disease
The protozoan Typanosomia cruzi causes Chagas disease, also known as American trypanosomiasis. The disease is transmitted through contact with the faeces/urine of infected triatomine bugs, (often known as kissing bugs), and blood transfusion. Twenty to thirty percent of infected people will develop cardiovascular, gastrointestinal, and/or neurological complications [84]. CD is endemic in Latin American countries, impacting 6–7 million people on a long-term basis and resulting in 14 thousand annual deaths. Moreover, due to international migration patterns, Chagas disease has now also becoming an issue in Canada, USA, several countries in Europe, and in Japan and Australia [26]. Nifurtimox and benznidazole are the only two drugs that are approved to treat chagas disease, since their introduction in the 60’s and 70’s [87]. Nevertheless, the safety and efficacy of both drugs are far from ideal. In most endemic Latin American nations, including Brazil, benznidazole is the only licensed medication. Since nifurtimox was taken off the market due to serious side effects and questions about its usefulness [26].
Leishmaniasis
Leishmaniasis threatens about 350 million people in 98 countries around the world. Several species of leishmania cause cutaneous, mucocutaneous, or visceral manifestations with variable degrees of severity. Cutaneous leishmania (CL) is the most common form, with an estimated 1.5 million cases per year. It is caused by L major, L. tropica, L. donovani, L. braziliensis, and other species. CL can range from a minor, self-limiting skin ulcer to a disfiguring and mutilating condition that can be fatal due to secondary consequences [26, 84].
Visceral Leishmaniasis (VL) or kala-azar is the most severe form of Leishmaniasis and is fatal if not treated. There are about 500 000 cases each year and mortality estimated at 10–20%, with more than 90% of these in India, Bangladesh, Brazil, Nepal, and Sudan. The current treatment for Leishmania have serious limitations, such as high cost, route of administration, side effects, and increased resistance [84]. Miltefosine is the only oral anti-leishmanial drug available in clinical practice for VL [88].
Compounds containing the quinolone scaffold are recently being reported for activity against the causal agents of these diseases. For example, Pedron and colleagues reported on the synthesis and antitrypanosomal activities of novel quinolone. These derivatives were evaluated in vitro for inhibitory activity against L. infantum, T. b. brucei, and T. cruzi. They were also investigated for cytotoxicity against HepG2 cell line. Compound 81, the lead compound from this study exhibited EC50 values of 12 and 500 nM against T.b. brucei trypomastigotes and T. cruzi amastigotes, respectively (see Fig. 7). In addition, the lead compound displayed moderate microsomal stability (half-life < 40 min) and favourable pharmacokinetic properties in mouse. Finally, it was shown to be bio-activated by type1 nitroreductase in both Leishmania and Trypanosoma [85].

Quinolones having activity against the trypanosoma and leishmania parasites
Hiltenspenger and colleagues reported a library of quinolones exhibiting potent antitrypanosomal activity. The lead compound (82) from this study however suffers from poor aqueous solubility [89].
Beteck et al. reported the synthesis and biological evaluation of a library of nonfluoroquinolones which exhibited moderate to poor antitrypanosomal activity, with the most active compound (83a) having an IC50 of 7.14 µM. Compounds in this study were also not cytotoxic [90]. In continuation of this work, Angula et al., investigated novel quinolone derivatives bearing heterorylidene moiety at position 6 of the quinolone ring for antitrypanosomal activity and identified compound 83b, having an IC50 value of 80 nM against T.b. brucei [91].
Motivated by the discovery of quinolone alkaloids (84) showing antiparasitic activity against leishmania sp. Suarez et al., investigated alkaloids isolated from the leaves of Raputia heptaphylla for antiparasitic activity against leishmania promastigotes and amastigotes. Two of the new compounds and the reference compound were effective against intracellular promastigotes. The two compounds (natural and synthetic) 85 were effective against intracellular promastigotes with EC50 values ranging from 1.6 to 9.9 μg/mL and amastigotes with EC50 ranging from 1.91 to 7.94 μg/mL. These alkaloids mode of action involves immune modulation through NO or ROS production which causes damage to the parasite’s cell wall and induction of apoptosis in infected cells [92].
Quinolone exhibiting antitrypanosomal activity has also been isolated from the root of waltheria indica. Zdorichenko et al., established the structure of waltherion F3 using X-ray crystallographic. Their synthetic strategy which utilised the Conrad-Limpach synthesis was utilised to achieved total synthesis of this compound and it will promote medicinal chemistry campaigns that focus on analogue synthesis [93].
Cretton et al. isolated 10 quinolone alkaloids (see Fig. 8) from the root extract of Waltheria indica. They characterised these compounds and evaluated them for antitrypanosomal activity against Trypanosoma cruzi. These compounds exhibited potent antitrypanosomal activity, exhibiting IC50 values in the range of 0.02–0.04 µM [94].

Quinolone alkaloids from extracts of Waltheria indica
When Furet & Pechere reviewed the improved antimicrobial activity of new fluoroquinolones and novel applications found for the drugs already marketed, it was discovered that pefloxacin, ofloxacin, and ciprofloxacin demonstrated antiprotozoal activity [95].
Quinolones active against cystic fibrosis (CF)
CF is a genetic disorder that severely affects vital organs such as the lungs, and digestive system and it is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) [96]. Thus, to better this dysfunction, drugs are needed, and one such drug is ivacaftor (88), a 4-quinolone-3-carboxamide based drug. Ivacaftor is currently the sole drug approved for CT. It works by increasing an open state for CFTR, thus increasing the activity of this channel [97]. Ivacaftor is used either alone or in combination with compounds [98] (see Fig. 9).

The structure of Ivacaftor
Vasudevan et al. reported a multi-step synthetic protocol for ivacaftor that allows for the synthesis of up to 7.2 g of the drug per day on a laboratory scale [98].
Guimbeilot et al. developed assays to determine the concentration of ivacaftor in cells and plasma. They observed that this drug has higher concentrations in cells than in plasma [97].
Rezniko et al. investigated ivacaftor for antibacterial activity. Because the structure of ivacaftor bears a quinolone ring and quinolones are known to be good antibiotics, Rezniko and colleagues were thus motivated to test ivacaftor against several bacteria. They observed that ivacaftor decreased the number of colonies forming units against Staphylococcus aureus and P. aeruginosen—and this effect was dose-dependent. Furthermore, ivacaftor was also observed to inhibit the growth of S.aureus and S. pneumonia [96].
Quinolones on cancer
Cancer is a complex disease which has abnormal cell growth as its hall mark. It arises from complex and numerous physiological changes within a cell, which ultimately lead to malignant tumours [99]. Many people may have tiny tumours called in situ tumours without being aware of them. For example, autopsy study on women between the ages of 40–50 years, who were not diagnosed with cancer before, found that one-third of this women had in situ tumours. Moreover, similar observations have been reported for men diagnosed with cancer. Although only 0.1% of men between the ages of 50–70 years are confirmed to have thyroid cancer, in situ carcinomas were detected in the thyroid glands of all autopsied men in this age group [100]. Although Richard Nixon declared war on cancer several decades ago, this disease is still a public health challenge presently, and research efforts on cures or management strategies for cancer are needed.
Voreloxin (89) is an anticancer agent analogous to quinolones [3]. It inhibits topoisomerase II, leading to DNA with abnormal structural integrity and ultimately cell apoptosis [101]. In their goal to discover quinolones with a mode of anticancer activity different from known anticancer agents, Tomita and co-workers identified the anticancer activity of voreloxin after evaluating several antibacterial quinolones and structurally related compounds for anticancer activity. They studied the analogues of 1,8-naphthyridine, investigated their structure–activity relationship and identified naphthyridine derivatives with IC50 values of 2.1 or 7.9 mg/mL against murine P 388 Leukaemia cells. Furthermore, derivatives with the naphthyridine core were the most active. Voreloxin had potent activity against eukaryotic cancer cell line, cytarabine and voreloxin synergize each other against acute myloid leukemia cancer cell lines and they both showed additive effect in a mouse model of bone marrow ablation [102]. Clinical evaluation of voreloxin shows that this compound has acceptable safety profile, pharmacokinetic parameters and maintain activity in patients [103].
Kljun et al. synthesised quinolone-ruthenium complexes and investigated their atitumour properties. They observed that these complexes demonstrated atitumour activity via DNA intercalation. Cathepsins B and S, which have been implicated in the development and progression of cancers, were also highlighted as possible targets for these complexes. They also investigated ruthenium-ofloxacin complex and observed that it alters DNA tertiary structure and is effective against the CH1 ovarian cancer cells [104].
Curcumin, a naturally occurring agent isolated from curcuma longa plant, has myriad pharmacological properties in vitro, and in vivo. Raghavan et al., synthesised several curcumin-quinolone hybrids and investigated their anticancer activity against SKOV3 cells. They observed that these compounds induced ROS and blebs formation in SKOV3 cells. SARs analyses in this study suggests that hybrids containing seven carbon linker had better cytotoxicity than hybrid containing five carbon [105].
Another novel discovery was the development of elacridar (90), which is a multidrug-resistant reversal agent. This molecule makes drug-resistant tumour cells susceptible to previously not effective chemotherapeutics. This compound is a quinolone derivative belonging to a class of organic compounds called the acridones [106]. Elacridar works by inhibiting P glycoprotein (P-gp) and breast cancer resistance protein (BCRP). The blood brain barrier (BBB) prevents substances, including drugs from entering the brain and uses efflux transporters, such as P-gp and BCRP to shield the brain [107]. Inhibition of these efflux transporters might be use to enhance drug permeation across the blood brain barrier, hence the discovery of elacridar is of such great importance. In addition, the oral bioavailability of drugs such as topotecan and paclitaxel have been increased using elacridar. Topotecan and paclitaxel are substrates for P-gp and BCRP [106]. Kuppens et al., observed that 100 mg of elacridar led to maximum oral bioavailability of topotecan [108].
FSL-61 (91), a 6-nitroquinolone, is another novel discovery. It serves as a fluorogenic probe for one electron reductases in hypoxic cells. One electron reductases are poorly characterised and there is a need to evaluate their activity in cells. Hypoxia is an important contributor to tumour progression. It has also been implicated in cancer treatment failure using radiotherapy and some chemotherapy. Su et al. identified, and characterised FSL-61 as one-electron reductase fluorogenic probe in hypoxic cell [109].
In another study by Green et al., FSL-61 was used to identify MsuE from P. aeruginosa as a novel and potential drug target that can be exploited using pro-drug strategy. The SOS and bacterial delivered enzyme prodrug cytotoxicity assays, however, showed that MsuE was not as appealing as nitroreductases from E. coli [110].
A study by Ceylane et al. investigated the antiproliferative effects of quinolones appended Schiff, and Mannich bases using the MTT method. The compounds were screened against 8 different human cancer cell lines. The results displayed that Mannich base derivatives have severe antiproliferative activity against prostate cancer (PC-3) ranging from 46.09% to 56.16% (when compared to imatinib mesylate) respectively. Furthermore, their antiproliferative activity was more effective than etoposide. Compound 92 showed the best antiproliferative activity against three cancer cell lines: exhibiting cell growth inhibition of 56.16% for prostate adenocarcinoma cell line (PC-3), 46.16% for hepatocellular carcinoma cell line (Hep3B), and 41.72% for breast cancer cell line (MCF-7). These compounds did not have a cytotoxic effect on normal cell [111] (see Fig. 10).

The structure of quinolones with activity against cancer
Quinolones on other diseases
Identification of monoamine oxidase (MAO) inhibitors is crucial in drug discovery against Alzheimer’s, Parkinson’s diseases, and depression. Studies reveal that quinolone isolated from the fruits of Evodia species inhibit leukotriene biosynthesis, activated T cells (NFAT), angiotensin II receptor binding and diacylglycerol acyltransferase activities [112, 113]. Han et al., carried out activity guided isolation and identified five quinolone alkaloids as MAO inhibitors. These compounds were more active against MAO-B than MAO-A [113].
Grover and Kini investigated a series of new nalidixic acid derivatives bearing quinazolinone moiety and identified some quinolone derivatives to be active against Streptococcus pygenes, candida albicans and Proteus vulgaris [114].
Ebola virus disease (EVD) is a severe and frequently lethal disease caused by Ebola virus (EBOV). EBOV has emerged as a significant public health concern between 2013 and 2016 in West Africa, claiming over 11,000 lives from a total of 30,000 cases that were recorded. In addition, in August 2018, 39,000 cases and more than 2200 deaths attributed to EVD occurred in the Democratic republic of Congo. EBOV was discovered in 1976 in the Democratic Republic of Congo and was the best known and studied member of the Filoviridae family long before the unprecedented West African outbreak [115]. EVD outbreak starts from a single case of probable zoonotic transmission, followed by human-to-human transmission either from direct contact with infected bodily fluids or fomites. It is characterized by fever and multiple organ dysfunction syndrome and has a high case fatality rate [116]. Gong et. al., evaluated the antiviral potencies of a panel of quinolones against EBOV in vitro and identified that Ryl-634, 93 and its derivatives were effective against EBOV at low nanomolar IC50 values and relatively high selectivity index. The candidate with the best activity was RYL-687, 94, exhibiting a low IC50 value of approximately 7 nM and was almost 6 times more potent than remdesivir (a monophosphoramidate prodrug of an adenosine analogue). Of note, the exogenous addition of different metabolites in the pyrimidine de novo synthesis pathway showed dihydroorotate dehydrogenase as the target of RYL-687. Thus, it is evident that such quinolone-derived compounds are promising therapeutic candidates against EBOV infection [117] (see Fig. 11).

Quinolone derivatives with ant-viral activity against EBOV
It has been hypothesised that commonly used pharmaceutical drugs cause persistent epigenetic changes. The word “epigenetic” refers to DNA and chromatin alterations that survive from one cell division to the next despite the underlying DNA sequences being unchanged [118]. Not only throughout development, but also throughout life, the epigenome (a cell's overall epigenetic state) is dynamic and receptive to environmental inputs [119]. Now, it is increasingly apparent that chemicals can cause changes in gene expression that persist long after exposure has ceased. Drugs may alter epigenetic homeostasis directly by affecting chromatin architecture or DNA methylation [120]. It is known that fluoroquinolones inhibit the bacterial DNA gyrase or the topoisomerase IV enzyme, thereby inhibiting DNA replication and transcription. Eukaryotic cells do not contain DNA gyrase or topoisomerase IV, so it has been assumed that fluoroquinolones have no effect on human cells, but they have been shown to inhibit eukaryotic DNA polymerase alpha and beta, and terminal deoxynucleotidyl transferase, affect cell cycle progression and function of lymphocytes in vitro, and cause other genotoxic effects [121]. Moreover, these agents have been associated with a diverse array of side-effects and interestingly, extensive changes in gene expression were found in articular cartilage of rats receiving the fluoroquinolone, ofloxacin, suggesting a potential epigenetic mechanism for the arthropathy caused by these agents. It has also been documented that the incidence of hepatic and dysrhythmic cardiovascular events following use of fluoroquinolones is increased compared to controls, suggesting the possibility of persistent gene expression changes in the liver and heart [121].
Conclusion and perspective
The development of quinolones started years ago and gained momentum around the 1970s as indicated by literature. Up to date, there still exist continues interest on quinolone derivatives; new compounds with improved activity are being discovered, new applications for quinolones already in the market are being found, and new methods to circumvent quinolone limitations are being developed. Thus, it is evident enough that the quinolone pharmacophore is an excellent and most versatile template for identification of new agents. Moreover, compounds containing the quinolone scaffold exhibit numerous pharmacological activities which include antibacterial, antimalarial, antiviral, antitrypanosomal, anticancer activities, MAO inhibitor, transmembrane conductance potentiator, fluorogenic probe. They exhibit numerous modes of actions which varies with the clinical condition and/or the chemistry around the quinolone nucleus; some reported targets for quinolone derivatives include topoisomerase iv, DNA gyrase, cytochrome bc1 complex, DprE1, nitro reductase, dihydrofolate reductase, NDH, P-gp. It is therefore apparent that the utility of quinolone derivatives extends far beyond the profile presented by fluoroquinolones (the most reported quinolone derivatives that have topoisomerase iv and DNA gyrase as targets) and that the quinolone scaffold, based in parts by the numerous pharmacological applications and the many well established synthetic protocols, is a viable lead source against any clinical condition.
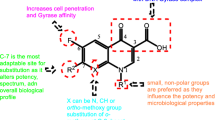
Since the historical moment of the discovery of nalidixic acid, the fluoroquinolone group of antibacterial have attracted a great deal of interest and attention from both the clinical and scientific research communities. Although a few representatives have proven their therapeutical value over time, many were withdrawn due to severe adverse reactions [122]. All fluoroquinolones form chelate complexes with divalent and trivalent cations. Fluoroquinolones' affinity for metal ions (Cu2 + > Fe2 + > Zn2 + > Mg2 + > Ca2 +) appears to be a necessary condition for their antibacterial activity wherein they bind to the DNA gyrase-complex via a magnesium ion. Furthermore, experimental evidence suggests that fluoroquinolones associated toxicities are based on their ability to chelate ions, most of which are co-factors, needed for proper functioning of human enzymes. For example, fluoroquinolones chondrotoxicity and mitochondria (a bacterial-like cell) damage are linked to magnesium-chelating characteristics [123]. The 3-carboxyl moiety ortho to the 4-oxo unit forms an optimal handle to chelate metal ions. Therefore, the structure of fluoroquinolones directly contributes to both their antibacterial activity and side effect profiles. Taking all these into consideration, it is our opinion that quinolone derivatives lacking the potential to chelate metal ions could be safer options to fluoroquinolones and that toxicities reported for fluoroquinolones should not be extrapolated to other quinolones without experimental evidence.
References
Dhiman P et al (2019) Recent advances in the synthetic and medicinal perspective of quinolones: a review. Bioorg Chem 92:103291. https://doi.org/10.1016/j.bioorg.2019.103291
Marganakop SB et al (2012) An efficient one-pot cyclization of quinoline thiosemicarbazones to quinolines derivatized with 1, 3, 4-thiadiazole as anticancer and anti-tubercular agents. Med Chem Res 21(2):185–191
Daneshtalab M, Ahmed A (2012) Nonclassical biological activities of quinolone derivatives. J Pharm Pharm Sci 15(1):52–72. https://doi.org/10.18433/J3302N
Lesher GY et al (1962) 1, 8-Naphthyridine derivatives. A new class of chemotherapeutic agents. J Med Chem 5(5):1063–1065
Andersson MI, MacGowan AP (2003) Development of the quinolones. J Antimicrob Chemother 51(suppl_1):1–11. https://doi.org/10.1093/jac/dkg212
Emmerson A, Jones A (2003) The quinolones: decades of development and use. J Antimicrob Chemother 51(suppl_1):13–20. https://doi.org/10.1093/jac/dkg208
Kumar KK et al (2011) Synthesis of quinoline coupled [1, 2, 3]-triazoles as a promising class of anti-tuberculosis agents. Carbohydr Res 346(14):2084–2090. https://doi.org/10.1016/j.carres.2011.06.028
Emami S, Shafiei A, Foroumadi A (2005) Quinolones: recent structural and clinical developments. IJPR 3:123–136
Marella A et al (2013) Quinoline: a versatile heterocyclic. Saudi Pharm J 21(1):1–12. https://doi.org/10.1016/j.jsps.2012.03.002
Pham TD, Ziora ZM, Blaskovich MA (2019) Quinolone antibiotics. Medchemcomm 10(10):1719–1739. https://doi.org/10.1039/C9MD00120D
Asif M (2013) Antirnicrobial and anti-tubercular activity of quinolone analogues. Sci Int 1(10):336–349. https://doi.org/10.17311/sciintl.2013.336.349
Netto Batalha P et al (2016) Quinolones in the search for new anticancer agents. Curr Pharm Des 22(39):6009–6020
Shi P et al (2017) Co (III)-catalyzed enaminone-directed C–H amidation for quinolone synthesis. Org Lett 19(9):2418–2421. https://doi.org/10.1021/acs.orglett.7b00968
Kaur N (2015) Benign approaches for the microwave-assisted synthesis of five-membered 1, 2-N, N-heterocycles. J Heterocycl Chem 52(4):953–973. https://doi.org/10.1002/jhet.2129
Duarte PD, Paixão MW, Corrêa AG (2013) Microwave assisted synthesis of 4-quinolones and N, Nʹ-diarylureas. Green Process Synth 2(1):19. https://doi.org/10.1515/gps-2012-0083
Zhao T, Xu B (2010) Palladium-catalyzed tandem amination reaction for the synthesis of 4-quinolones. Org Lett 12(2):212–215. https://doi.org/10.1021/ol902626d
Shao J et al (2012) Synthesis of N-alkyl-substituted 4-quinolones via tandem alkenyl and aryl C–N bond formation. ChemInform 43(41):1798. https://doi.org/10.1055/s-0031-1290775
Xu X, Zhang X (2017) Direct synthesis of 4-quinolones via copper-catalyzed anilines and alkynes. Org Lett 19(18):4984–4987. https://doi.org/10.1021/acs.orglett.7b02495
Åkerbladh L et al (2015) Synthesis of 4-quinolones via a carbonylative sonogashira cross-coupling using molybdenum hexacarbonyl as a CO source. J Org Chem 80(3):1464–1471. https://doi.org/10.1021/jo502400h
Hu W et al (2015) Direct synthesis of 2-aryl-4-quinolones via transition-metal-free intramolecular oxidative C (sp3)–H/C (sp3)–H coupling. Org Lett 17(5):1268–1271. https://doi.org/10.1021/acs.orglett.5b00248
Kang D, Hong S (2015) Rh (III) and Ru (II)-catalyzed site-selective C–H alkynylation of quinolones. Orga Lett 17(8):1938–1941. https://doi.org/10.1021/acs.orglett.5b00641
Saito K, Moriya Y, Akiyama T (2015) Chiral phosphoric acid catalyzed asymmetric synthesis of 2-substituted 2, 3-dihydro-4-quinolones by a protecting-group-free approach. Org Lett 17(13):3202–3205. https://doi.org/10.1021/acs.orglett.5b01654
Monastyrskyi A, Namelikonda NK, Manetsch R (2015) Metal-free arylation of ethyl acetoacetate with hypervalent diaryliodonium salts: an immediate access to diverse 3-aryl-4 (1 H)-quinolones. J Org Chem 80(5):2513–2520. https://doi.org/10.1021/jo5023958
Wang W et al (2015) Synthesis of quinolinones with palladium-catalyzed oxidative annulation between acrylamides and arynes. J Org Chem 80(5):2835–2841. https://doi.org/10.1021/jo5027673
Manikandan R, Jeganmohan M (2014) Ruthenium-catalyzed cyclization of anilides with substituted propiolates or acrylates: an efficient route to 2-quinolinones. Org Lett 16(13):3568–3571. https://doi.org/10.1021/ol501548e
Sangenito LS et al (2019) Leishmaniasis and Chagas disease–neglected tropical diseases: treatment updates. Curr Top Med Chem 19(3):174–177
WHO (2020) World malaria report 2020: 20 years of global progress and challenges. ISBN 978-92-4-001579-1 (electronic version). https://apps.who.int/iris/handle/10665/337660 Accessed 27 May 2022
Mbengue A et al (2015) A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520(7549):683–687
Ashley EA et al (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N Eng J Med 371(5):411–423. https://doi.org/10.1056/NEJMoa1314981
Balogun TA, Omoboyowa DA, Saibu OA (2020) In silico anti-malaria activity of quinolone compounds against Plasmodium falciparum dihydrofolate reductase (pfDHFR). Int J Biochem Res Rev 29(8):10–17. https://doi.org/10.9734/ijbcrr/2020/v29i830208
Beteck RM et al (2014) Recent progress in the development of anti-malarial quinolones. Malar J 13(1):1–10
Van Schalkwyk DA et al (2020) Novel endochin-like quinolones exhibit potent in vitro activity against Plasmodium knowlesi but do not synergize with proguanil. Antimicrob Agents Chemother 64(5):e02549-e2619. https://doi.org/10.1128/AAC.02549-19
Doggett JS et al (2020) Orally bioavailable endochin-like quinolone carbonate ester prodrug reduces Toxoplasma gondii brain cysts. Antimicrob Agents Chemother 64(9):e00535-e620. https://doi.org/10.1128/AAC.00535-20
Pou S et al (2021) New scalable synthetic routes to ELQ-300, ELQ-316, and other antiparasitic quinolones. Org Process Res Dev 25(8):1841–1852. https://doi.org/10.1021/acs.oprd.1c00099
Nilsen A et al (2013) Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 5(177):177ra37-177ra37. https://doi.org/10.1126/scitranslmed.30050
Miley GP et al (2015) ELQ-300 prodrugs for enhanced delivery and single-dose cure of malaria. Antimicro Agents Chemother 59(9):5555–5560. https://doi.org/10.1128/AAC.01183-15
Frueh L et al (2017) Alkoxycarbonate ester prodrugs of preclinical drug candidate ELQ-300 for prophylaxis and treatment of malaria. ACS Infect Dis 3(10):728–735. https://doi.org/10.1021/acsinfecdis.7b00062
Garrido-Cardenas JA, Mesa-Valle C, Manzano-Agugliaro F (2018) Human parasitology worldwide research. Parasitology 145(6):699–712. https://doi.org/10.1017/S0031182017001718
Balbino LS et al (2021) Epidemiological study of toxoplasmosis outbreaks in Brazil. Transbound Emerg Dis. https://doi.org/10.1111/tbed.14214
Hlaváčová J et al (2021) Male-to-female presumed transmission of toxoplasmosis between sexual partners. Am J Epidemiol 190(3):386–392. https://doi.org/10.1093/aje/kwaa198
Eberhard N et al (2020) Activities of endochin-like quinolones against in vitro cultured Besnoitia besnoiti tachyzoites. Front Vet Sci 7:96. https://doi.org/10.3389/fvets.2020.00096
Ryley J, Peters W (1970) The antimalarial activity of some quinolone esters. Ann Trop Med Parasitol 64(2):209–222. https://doi.org/10.1080/00034983.1970.11686683
Cross RM et al (2011) Synthesis, antimalarial activity, and structure–activity relationship of 7-(2-Phenoxyethoxy)-4 (1 H)-quinolones. J Med Chem 54(24):8321–8327. https://doi.org/10.1021/jm200718m
Neelarapu R et al (2018) Design and synthesis of orally bioavailable piperazine substituted 4 (1 h)-quinolones with potent antimalarial activity: structure–activity and structure–property relationship studies. J Med Chem 61(4):1450–1473. https://doi.org/10.1021/acs.jmedchem.7b00738
Cowley R et al (2012) The development of quinolone esters as novel antimalarial agents targeting the Plasmodium falciparum bc 1 protein complex. MedChemComm 3(1):39–44. https://doi.org/10.1039/C1MD00183C
Nam T-G et al (2011) A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol 6(11):1214–1222. https://doi.org/10.1021/cb200105d
Beteck RM et al (2018) Accessible and distinct decoquinate derivatives active against Mycobacterium tuberculosis and apicomplexan parasites. Commun Chem 1(1):1–11
Beteck RM et al (2016) Straightforward conversion of decoquinate into inexpensive tractable new derivatives with significant antimalarial activities. Bioorg Med Chem Lett 26(13):3006–3009. https://doi.org/10.1016/j.bmcl.2016.05.024
Leung SC et al (2012) Identification, design and biological evaluation of heterocyclic quinolones targeting Plasmodium falciparum type II NADH: quinone oxidoreductase (PfNDH2). J Med Chem 55(5):1844–1857. https://doi.org/10.1021/jm201184h
Ginsburg AS, Grosset JH, Bishai WR (2003) Fluoroquinolones, tuberculosis, and resistance. Lancet Infect Dis 3(7):432–442. https://doi.org/10.1016/S1473-3099(03)00671-6
Dube PS et al (2021) Easily accessed nitroquinolones exhibiting potent and selective anti-tubercular activity. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2021.113207
WHO (2021) WHO global lists of high burden countries for tuberculosis (TB), TB/HIV and multidrug/rifampicin-resistant TB (MDR/RR-TB), 2021–2025: background document. https://apps.who.int/iris/handle/10665/341980. Accessed 27 May 2022
Grant AD, De Cock KM (2001) HIV infection and AIDS in the developing world. BMJ 322(7300):1475–1478. https://doi.org/10.1136/bmj.322.7300.1475
Wang R, Xu K, Shi W (2019) Quinolone derivatives: potential anti-HIV agent—development and application. Arch Pharm 352(9):1900045. https://doi.org/10.1002/ardp.201900045
Marchetti G, Tincati C, Silvestri G (2013) Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev 26(1):2–18. https://doi.org/10.1128/CMR.00050-12
WHO (2020) Health policy and system support to optimize community health worker programmes for HIV, TB and malaria services: an evidence guide. https://apps.who.int/iris/handle/10665/340078. Accessed 27 May 2022
Abdool Karim SS et al (2010) Timing of initiation of antiretroviral drugs during tuberculosis therapy. N Eng J Med 362(8):697–706. https://doi.org/10.1056/NEJMoa0905848
Zhao X, Wang X, Li Y (2019) Combined HQSAR method and molecular docking study on genotoxicity mechanism of quinolones with higher genotoxicity. Environ Sci Pollut Res 26(34):34830–34853
Asif M (2015) Role of quinolones and quinoxaline derivatives in the advancement of treatment of tuberculosis. Int J Sci World 3:18–36. https://doi.org/10.14419/ijsw.v3i1.3432
Sulochana S, Rahman F, Paramasivan C (2005) In vitro activity of fluoroquinolones against Mycobacterium tuberculosis. J Chemother 17(2):169–173. https://doi.org/10.1179/joc.2005.17.2.169
Liu B et al (2018) quinoline derivatives with potential activity against multidrug-resistant tuberculosis. J Heterocyl Chem 55(8):1863–1873. https://doi.org/10.1002/jhet.3241
Nijland H et al (2007) Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin Infect Dis 45(8):1001–1007. https://doi.org/10.1086/521894
Senthilkumar P et al (2009) Synthesis and antimycobacterial activities of novel 6-nitroquinolone-3-carboxylic acids. Eur J Med Chem 44(1):345–358. https://doi.org/10.1016/j.ejmech.2008.02.031
Beteck RM et al (2019) new quinolone-based thiosemicarbazones showing activity against plasmodium falciparum and mycobacterium tuberculosis. Molecules. https://doi.org/10.3390/molecules24091740
Huang H et al (2005) Identification and active expression of the Mycobacterium tuberculosis gene encoding 5-phospho-α-D-ribose-1-diphosphate: decaprenyl-phosphate 5-phosphoribosyltransferase, the first enzyme committed to decaprenylphosphoryl-D-arabinose synthesis. J Biol Chem 280(26):24539–24543. https://doi.org/10.1074/jbc.M504068200
Naik M et al (2014) 4-Aminoquinolone piperidine amides: noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J Med Chem 57(12):5419–5434. https://doi.org/10.1021/jm5005978
Chong SMS et al (2020) Antituberculosis activity of the antimalaria cytochrome bcc oxidase inhibitor SCR0911. ACS Infect Dis 6(4):725–737. https://doi.org/10.1021/acsinfecdis.9b00408
Bahuguna A, Rawat S, Rawat DS (2021) QcrB in Mycobacterium tuberculosis: the new drug target of antitubercular agents. Med Res Rev. https://doi.org/10.1002/med.21779
Hong WD et al (2017) Rational design, synthesis, and biological evaluation of heterocyclic quinolones targeting the respiratory chain of Mycobacterium tuberculosis. J Med Chem 60(9):3703–3726. https://doi.org/10.1021/acs.jmedchem.6b01718
Marinello J et al (2008) Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochem 47(36):9345–9354. https://doi.org/10.1021/bi800791q
Deeks ED (2014) Elvitegravir: a review of its use in adults with HIV-1 infection. Drugs 74(6):687–697
Shimura K, Kodama EN (2009) Elvitegravir: a new HIV integrase inhibitor. Antivir Chem Chemother 20(2):79–85. https://doi.org/10.3851/IMP1397
He Q-Q et al (2011) Synthesis and biological evaluation of HQCAs with aryl or benzyl substituents on N-1 position as potential HIV-1 integrase inhibitors. Bioorg Med Chem 19(18):5553–5558. https://doi.org/10.1016/j.bmc.2011.07.037
He Q-Q et al (2013) Synthesis and biological evaluation of 5-fluoroquinolone-3-carboxylic acids as potential HIV-1 integrase inhibitors. J Enzyme Inhib Med Chem 28(4):671–676. https://doi.org/10.3109/14756366.2012.668540
Curreli F et al (2011) Virtual screening based identification of novel small-molecule inhibitors targeted to the HIV-1 capsid. Bioorg Med Chem 19(1):77–90. https://doi.org/10.1016/j.bmc.2010.11.045
Costi R et al (2014) Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J Med Chem 57(8):3223–3234. https://doi.org/10.1021/jm5001503
Kashiwase H et al (1999) A new fluoroquinolone derivative exhibits inhibitory activity against human immunodeficiency virus type 1 replication. Chemotherapy 45(1):48–55. https://doi.org/10.1159/000007164
Hagihara M et al (1999) Synthesis and anti-HIV activity of arylpiperazinyl fluoroquinolones: a new class of anti-HIV agents. Bioorg Med Chem Lett 9(21):3063–3068. https://doi.org/10.1016/S0960-894X(99)00537-5
Yoneda JD et al (2014) Docking of anti-HIV-1 oxoquinoline-acylhydrazone derivatives as potential HSV-1 DNA polymerase inhibitors. J Mol Struct 1074:263–270. https://doi.org/10.1016/j.molstruc.2014.05.081
Jadulco RC et al (2014) 4-Quinolone alkaloids from melochia odorata. J Nat Prod 77(1):183–187. https://doi.org/10.1021/np400847t
Massari S et al (2009) Studies on anti-HIV quinolones: new insights on the C-6 position. Bioorg Med Chem 17(2):667–674. https://doi.org/10.1016/j.bmc.2008.11.056
Cecchetti V et al (2000) 6-Aminoquinolones as new potential anti-HIV agents. J Med Chem 43(20):3799–3802. https://doi.org/10.1021/jm9903390
Parolin C et al (2003) New anti-human immunodeficiency virus type 1 6-aminoquinolones: mechanism of action. Antimicrob Agents Chemother 47(3):889–896. https://doi.org/10.1128/AAC.47.3.889-896.2003
WHO (2012) Research priorities for Chagas disease, human African trypanosomiasis and leishmaniasis. World Health Organization
Pedron J et al (2020) New 8-nitroquinolinone derivative displaying submicromolar in vitro activities against both Trypanosoma brucei and cruzi. ACS Med Chem Lett 11(4):464–472. https://doi.org/10.1021/acsmedchemlett.9b00566
Berninger M et al (2017) Novel lead compounds in pre-clinical development against African sleeping sickness. MedChemComm 8(10):1872–1890. https://doi.org/10.1039/C7MD00280G
Croft SL, Barrett MP, Urbina JA (2005) Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol 21(11):508–512. https://doi.org/10.1016/j.pt.2005.08.026
Khare S et al (2016) Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537(7619):229–233
Hiltensperger G et al (2012) Synthesis and structure–activity relationships of new quinolone-type molecules against Trypanosoma brucei. J Med Chem 55(6):2538–2548. https://doi.org/10.1021/jm101439s
Beteck RM et al (2018) Synthesis, in vitro cytotoxicity and trypanocidal evaluation of novel 1, 3, 6-substituted non-fluoroquinolones. S Afr J Chem 71:188–195. https://doi.org/10.17159/0379-4350/2018/v71a25
Angula KT et al (2022) Synthesis and in vitro antitrypanosomal evaluation of novel 6-heteroarylidene-substituted quinolone derivatives. Eur J Med Chem 227:113913
Torres Suarez E et al (2020) Antileishmanial activity of synthetic analogs of the naturally occurring quinolone alkaloid N-methyl-8-methoxyflindersin. PLoS ONE 15(12):e0243392
Zdorichenko V et al (2019) The Synthesis of waltherione f and its analogues with modifications at the 2-and 3-positions as potential antitrypanosomal agents. Chem Eur J 25(5):1286–1292. https://doi.org/10.1371/journal.pone.0243392
Cretton S et al (2014) Antitrypanosomal quinoline alkaloids from the roots of Waltheria indica. J Nat Prod 77(10):2304–2311. https://doi.org/10.1021/np5006554
Furet Y, Pechere J (1991) Newly documented antimicrobial activity of quinolones. Eur J Clin microbiol infect Dis 10(4):249–254
Reznikov LR et al (2014) Antibacterial properties of the CFTR potentiator ivacaftor. J Cyst Fibros 13(5):515–519. https://doi.org/10.1016/j.jcf.2014.02.004
Guimbellot JS et al (2020) Variable cellular ivacaftor concentrations in people with cystic fibrosis on modulator therapy. J Cyst Fibros 19(5):742–745. https://doi.org/10.1016/j.jcf.2020.01.011
Vasudevan N et al (2018) A multi-step continuous flow synthesis of the cystic fibrosis medicine ivacaftor. React Chem Eng 3(4):520–526
Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab 7(1):1–22
Folkman J, Kalluri R (2004) Cancer without disease. Nature 427(6977):787–787
Hawtin RE et al (2010) Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS ONE 5(4):e10186. https://doi.org/10.1371/journal.pone.0010186
Tomita K et al (2002) Synthesis and structure− activity relationships of novel 7-substituted 1, 4-dihydro-4-oxo-1-(2-thiazolyl)-1, 8-naphthyridine-3-carboxylic acids as antitumor agents. Part 1. J Med Chem 45(25):5564–5575. https://doi.org/10.1021/jm010057b
Advani RH et al (2010) Voreloxin, a first-in-class anticancer quinolone derivative, in relapsed/refractory solid tumors: a report on two dosing schedules. Clin Cancer Res 16(7):2167–2175. https://doi.org/10.1158/1078-0432.CCR-09-2236
Kljun J et al (2013) New uses for old drugs: attempts to convert quinolone antibacterials into potential anticancer agents containing ruthenium. Inorg Chem 52(15):9039–9052. https://doi.org/10.1021/ic401220x
Raghavan S et al (2015) Synthesis and anticancer activity of novel curcumin–quinolone hybrids. Bioorg Med Chem Lett 25(17):3601–3605. https://doi.org/10.1016/j.bmcl.2015.06.068
Sane R, Mittapalli RK, Elmquist WF (2013) Development and evaluation of a novel microemulsion formulation of elacridar to improve its bioavailability. J Pharm Sci 102(4):1343–1354. https://doi.org/10.1002/jps.23450
Kallem R et al (2012) A simplified protocol employing elacridar in rodents: a screening model in drug discovery to assess P-gp mediated efflux at the blood brain barrier. Drug Metab Lett 6(2):134–144. https://doi.org/10.2174/187231212804096682
Kuppens IE et al (2007) A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res 13(11):3276–3285. https://doi.org/10.1158/1078-0432.CCR-06-2414
Su J, Guise CP, Wilson WR (2013) FSL-61 is a 6-nitroquinolone fluorogenic probe for one-electron reductases in hypoxic cells. Biochem J 452(1):79–86. https://doi.org/10.1042/BJ20121695
Green LK et al (2013) The flavin reductase MsuE is a novel nitroreductase that can efficiently activate two promising next-generation prodrugs for gene-directed enzyme prodrug therapy. Cancers 5(3):985–997. https://doi.org/10.3390/cancers5030985
Ceylan Ş et al (2020) antimicrobial, antioxidant and antiproliferative activities of novel quinolones. ChemistrySelect 5(36):11340–11346. https://doi.org/10.1002/slct.202002779
Lee MK et al (2003) 1-methyl-2-undecyl-4 (1H)-quinolone as an irreversible and selective inhibitor of type B monoamine oxidase. Chem Pharm Bull 51(4):409–411. https://doi.org/10.1248/cpb.51.409
Han XH et al (2007) Quinolone alkaloids from Evodiae fructus and their inhibitory effects on monoamine oxidase. Arch Pharm Res 30(4):397–401
Grover G, Kini SG (2006) Synthesis and evaluation of new quinazolone derivatives of nalidixic acid as potential antibacterial and antifungal agents. Eur J Med Chem 41(2):256–262. https://doi.org/10.1016/j.ejmech.2005.09.002
Feldmann H, Sprecher A, Geisbert TW (2020) Ebola. N Eng J Med 382(19):1832–1842. https://doi.org/10.1056/NEJMra1901594
Jacob ST et al (2020) Ebola virus disease. Nat Rev Dis Primers 6(1):1–31
Gong M et al (2021) Novel quinolone derivatives targeting human dihydroorotate dehydrogenase suppress Ebola virus infection in vitro. Antivir Res 194:105161. https://doi.org/10.1016/j.antiviral.2021.105161
McKay JA, Mathers JC (2011) Diet induced epigenetic changes and their implications for health. Acta Physiol 202(2):103–118. https://doi.org/10.1111/j.1748-1716.2011.02278.x
Kondo Y, Issa J-PJ (2004) Epigenetic changes in colorectal cancer. Cancer Metastasis Rev 23(1):29–39
Grønbaek K, Hother C, Jones PA (2007) Epigenetic changes in cancer. APMIS 115(10):1039–1059. https://doi.org/10.1111/j.1600-0463.2007.apm_636.xml.x
Csoka AB, Szyf M (2009) Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Med Hypotheses 73(5):770–780. https://doi.org/10.1016/j.mehy.2008.10.039
Rusu A et al (2021) Structural characterization of the millennial antibacterial (fluoro) quinolones—shaping the fifth generation. Pharmaceutics 13(8):1289. https://doi.org/10.3390/pharmaceutics13081289
Stahlmann R, Lode H (1998) Safety of quinolones. Lancet 352(9136):1313. https://doi.org/10.1016/S0140-6736(05)70526-2
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dube, P.S., Legoabe, L.J. & Beteck, R.M. Quinolone: a versatile therapeutic compound class. Mol Divers 27, 1501–1526 (2023). https://doi.org/10.1007/s11030-022-10581-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10581-8