
Abstract
Papain-like protease (nsp-3; non-structural protein) of novel corona virus is an ideal target for developing drugs as it plays multiple important functions for viral growth and replication. For instance, role of nsp-3 has been recognized in cleavage of viral polyprotein; furthermore, in infected host it weakens the immune system via downregulating the production of type I interferon. This downregulation is promoted by removal of ubiquitin-like interferon-stimulated gene 15 protein (ISG15) from interferon-responsive factor 3 (IRF3) protein. Among known inhibitors of SARS-CoV-PLpro GRL0617 is by far the most effective inhibitor. As PLpro of SARS-CoV2 is having more than 80% similarity with SARS-CoV-PLpro, GRL0617 is reported to be effective even against SARS-CoV2. Owing to this similarity, certain key amino acids remain the same/conserved in both proteins. Among conserved amino acids Tyr268 for SARS-CoV2 and Tyr269 for SARS-CoV produce important hydrophobic interactions with aromatic rings of GRL0617. Here, in this study antibacterial compounds were collected from ZINC database, and they were filtered to select compounds that are having similar structural features as GRL0617. This filtered library of compound was then docked with SARS-CoV and CoV2-PLpro. Five hits were noted that were able to interact with Tyr268 (SARS-CoV2) and Tyr269 (SARS-CoV). Further, best hit 2-(2-((benzofuran-2-carboxamido)methyl)-5-methoxy-1H-indol-1-yl)acetic acid (ZINC44459905) was studied using molecular dynamic simulation where stability of protein–ligand complex as well as stability of produced interactions was noted.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Globally, Severe Acute Respiratory Syndrome Virus-Coronavirus-2 (SARS-CoV-2) has infected nearly ~ 102 million people of which ~ 2 million have passed and yet the mayhem is not over. It has been a year since the first appearance of virus and the extremely infectious nature of virus are causing the community-level infection transmissions in nearly 137 countries (https://covid19.who.int/). Scientific communities and clinicians are attempting to develop ways to hinder the progress of virus via inhibiting the host-to-host transmissions or replications within host, but success is yet to be achieved [1,2,3,4]. Also, the classification of SARS-CoV-2, provided by International Committee on Taxonomy of Viruses (ICTV), reveals virus to be genetically related to SARS-CoV which was spotted in year 2003 [5,6,7]. Unlike SARS-CoV, SARS-CoV-2 is highly infectious and in a very brief amount of time, it has spread all over the world. Respiratory and gastrointestinal systems are the primary targets of the virus where establishment of infection is often followed by common flu-like symptoms such as fever, shortness of breath, pneumonia. However, the mortality rate of the infection is lower and most people that have passed were suffering from pre-existing medical conditions such as diabetes, heart disease. Regardless, virus has been reported to cause nervous system-related abnormalities in people that have recovered; hence, the full extent of the infection is yet hazy [8]. Such uncertain after effects warrant effective ways to control the spread of virus either by vaccine or drug that can inhibit or reduce the severity of infection. Development of vaccine has been the prime target for major pharmaceutical industries. Furthermore, strategies to develop antiviral drugs have been proposed that targets the developmental stages of virus. Propagation of virus requires specific set of structural and non-structural proteins. Absence of such proteins can restrict the host-to-host transfer or replication which ultimately results in inhibition of the virus spread. Hence, drugs capable of restricting the availability of these proteins are sought after [3]. In SARS virus, 12 such structural and non-structural targets have been identified. Among structural proteins Spike protein (S-protein), Envelop small membrane protein (E-protein), Membrane protein (M-protein), Nucleocapsid protein (N-protein), are mandatory for virus to attain its final virulent physical form, whereas non-structural proteins include Main protease (Mpro) (nsp-5), Papain-like protease (PLpro) (nsp-3), nsp-10, nsp-11 nsp-13, nsp-14, nsp-15 and nsp-16 that plays cardinal role in replication and genome packaging in coat protein [9]. Proteins require certain conformations to retain their specific abilities; mere misfolding can render the protein inactive and hinder the viral infection; hence, these proteins have been perceived as a target [5, 10].
Over the course of evolution cells have developed immune responses to fight against viral infections. Two such mechanisms that have been observed against number of viruses are ubiquitination and ISGylation. Ubiquitination is a mechanism developed by cells to undergo post-translational regulations on proteins where lysine residue of protein is loaded with short ubiquitin chains that are identified as a signal for proteasomal degradation of protein [11, 12]. In a similar manner ISGylation is also a process developed for regulation of target proteins. Here, IFN-1 induces the synthesis of interferon-stimulated gene 15 (ISG15) which in turn binds with the target protein to regulate its function. As both pathways are active against viral invasion, viruses have developed various tactics to hinder their function (Fig. 1). Gene nsp3 of CoV encodes the non-structural protein PLpro which is protease. Subsequently, this protease cleaves the nsp1, nsp2, and nsp3 in to separate proteins that are mandatory for transcription and replication of viral genome. PLpro encoded by both SARS-CoV and SARS-CoV2 is essentially similar in a manner of its function against innate viral defenses such as ubiquitination and ISGylation. However, both proteins differ in terms of their efficiencies due to changes in their active sites. For instance, SARS-CoV2 is better at deISGylating than deubiquitylating [13]. Even though process of ISG15 cleavage by SARS-CoV2-PLpro is not clearly understood, it has been proposed that interactions between Val66 and hydrophobic surface of the N-terminal ubiquitin fold domain of ISG15 are responsible factors, whereas, in SARS-CoV-PLpro, Leu76 uses hydrophobic interactions to promote the contact with Ile44 of ubiquitin. In SARS-CoV2, Leu is replaced by Thr which is having lower affinity for ubiquitin [13].

Illustration representing role of IRF3 in synthesis of INF1 and its inhibition by viral PLpro along with other cellular disturbances caused by viral entry and replication
PLpro promotes viral propagation via two simultaneous activities that are polyprotein cleavage and obstruction of host innate immune response. Such dual role of PLpro in propagation of virus and suppression of host immune response makes it ideal target for drugs development. One such drug, GRL0617, has been reported as an inhibitor of SARS-COV-PLpro. In vitro studies conducted on lung cancer line prove the efficiency of GRL0617 as a SARS-CoV2-PLpro inhibitor, not only that drug is also capable of IFN-1 response restoration in cells exposed to SARS-COV2-PLpro. Furthermore, replication of SARS-CoV2 has been reported to be effectively hindered by 100 μM of GRL0617 [13]. In-silico studies have shown that Tyr present at position 268 and 269 in SARS-CoV2-PLpro and SARS-CoV-PLpro, respectively, takes active part in interaction with naphthalene drug GRL0617. Furthermore, the PLpro encoded by the Middle East Respiratory Syndrome (MERS)-CoV has been reported to be immune against GRL0617 as the key conserved amino acid Tyr has been replaced by Thr which further proves the importance of Tyr268 as a target [13].
Lengthy time periods and research expenses for development of novel drugs have recently favored the idea of drug repurposing which revolves around the concept of using already known and established drugs to treat new diseases via targeting the overlapping pathways or applying as ligand analog or via targeting the proteins homologs. Drug repurposing approach was previously reported with sildenafil citrate for erectile dysfunction and thalidomide for erythema nodosum leprosum (ENL) and multiple myeloma, but these instances were based on serendipity. Recently, development of computer-aided drug discovery (CADD) methods such as molecular docking, molecular dynamics (MD), free energy perturbations (FEP), quantitative structure–activity relationship (QSAR) modeling, and knowledge-mining approaches, along with machine learning methods has enabled the tools required for active application of drug repurposing [14, 15]. In this study, concept of drug repurposing is applied on the library of antibacterial compounds obtained from Zinc database (ZINC (docking.org)) to identify the potential leads for SARS-CoV-PLpro inhibition using GRL0617 as template. Such approach will significantly reduce the time required for development and introduction of new drug in market as these compounds are already synthesized all the primary information of their effectivity is already reported. Here, molecular docking was used to screen the antibacterial compounds via studying and comparing their abilities to mimic the interactions produced by GRL0617. Screening was conducted based on following four abilities: (1) bond formation with Tyr268 (SARS-CoV2-PLpro) and Tyr269 (SARS-CoV-PLpro), (2) protein–ligand binding energy, (3) H– bond formation, (4) hydrophobic bond formation. Results of docking revealed five compounds capable of binding with Tyr268 and Tyr269. 2-(2-((Benzofuran-2-carboxamido)methyl)-5-methoxy-1H-indol-1-yl)acetic acid (ZINC44459905), ligand with highest binding efficiency, contains two ring structures benzofuran and indole that takes active part in creation of interactions similar to GRL0617 naphthalene moiety with Tyr268 and Tyr269. To further confirm the stability of interactions, molecular dynamics study of GRL0617 and ZINC44459905 was performed and compared. Furthermore, ADMET (Absorption–Distribution–Metabolism–Excretion–Toxicity) analysis of control and all the proposed compound was performed. The drug-like compound 2-(2-((benzofuran-2-carboxamido)methyl)-5-methoxy-1H-indol-1-yl)acetic acid (ZINC44459905) might be employed in the search for COVID-19 absolutely necessary treatment.
Materials and methods
PLpro sequence retrieval and comparison
After ensuring the similarities in their sequence and structure without any mutation, sequences of SARS-CoV and SARS-CoV2-PLpro were retrieved from PDB database using PDB ID’s 3E9S and 7CMD, respectively. Sequences were selected for docking only after ensuring the presence of Tyr at position 269 in 3E9S and 268 in 7CMD.
Receptor–ligand preparation
Both PLpro proteins of SARS-CoV and SARS-CoV2, 7CMD and 3E9S, respectively, are available on PDB in co-crystalized form with GRL0617. Prior to molecular docking, binding site for test ligands was determined to be the same as GRL0617. Hence, the XYZ co-ordinates for both proteins were identified. Furthermore, both proteins were prepared for docking by removing the all-bound ligands, ions and water molecules. Protein preparation wizard of maestro was used to assign hydrogen atoms, charges and to perform energy minimization of protein structure [16]. After pre-processing, protein was optimized using refine option in the wizard. Once optimization was performed, energy minimization of protein was performed using OPLS3 forcefield [17, 18]. Once protein was minimized, receptor grid generation wizard was used to generate the glide grid [19]. Test ligand for molecular docking was obtained from ZINC database where under the category of organism bacteria keyword was used to separate the compounds of interest. Prior to docking they were optimized via hydrogen atom addition and energy minimization in LigPrep wizard with OPLS3 forcefield. At last, minimized ligands were saved and used for docking analysis [20, 21].
Molecular ligand–receptor docking
Molecular docking analysis of proteins and ligands was carried out using ligand docking wizard [22] of Schrödinger Maestro software. ~ 8000 test ligands obtained from zinc database were docked in binding cavity of both target PLpro proteins that was previously occupied by GRL0617. Moreover, native ligand GRL0617 was used as a positive control. The optimized and minimized protein from the previous step was used for docking. The first step for docking is to prepare the grid at the exact same co-ordinates as that of native ligand. The grid box of the size 15 Å × 15 Å × 15 Å was prepared. The grid for docking with the mentioned parameters was prepared using ‘Receptor grid generation’ feature of Glide module in Schrödinger Maestro. For docking, the output file of (1) receptor grid generation and (2) prepared minimized ligands was imported in the ‘Ligand docking’ window of Glide module in Schrödinger Maestro. Under the settings, the precision of docking was set as ‘Extra Precision (XP),’ Ligand sampling was set as ‘Flexible,’ and the Epik state penalties were added to docking score. The output was set to show only the best pose. The entire docking was performed using Glide module in Schrödinger Maestro [23, 24]. The entire output file of docking was used for performing MM-GBSA assessment.
To perform docking, in ligand docking wizard, key parameters were kept as following: (1) precision: high-throughput virtual screening; (2) ligand sampling: flexible; and (3) ligands with more than 500 atoms and 100 rotatable bonds were ignored. Top hits of high-throughput virtual screening were manually analyzed for their structural similarities with GRL0617 and interactions, and ideal matches were screened. Screened compounds were docked using extra precision option in the wizard. After docking single pose with minimum binding energy (kcal/mol) selected for further studies. After the selection of pose, each ligand was opened in BIOVIA Discovery Studio (DS) visualizer for analysis of interactions taking place between ligand and amino acids of protein. To narrow the search for ideal ligand, test ligands were filtered based on their ability to interact with Tyr268 and Tyr269 of SARS-CoV2-PLpro and SARS-CoV-PLpro, respectively. Ligand capable of producing maximum interactions like GRL0617, with both target proteins 7CMD and 3E9S, was selected for molecular dynamics simulation analysis where stability of ligand–receptor complex was validated.
MM-GBSA calculation
Molecular mechanics generalized born surface area (MM-GBSA) calculation was used to calculate the binding free energy change [25,26,27,28]. Docked ligand–receptor complexes were minimized using local optimization feature present in Prime wizard of Maestro (Schrödinger Release 2017-4). Binding energy for each receptor–ligand complex was determined by using OPLS-2005 force field. Equation employed for free energy calculation is as follows:
Here ΔEMM represents the variation between the minimized energy of the receptor–ligand complexes; ΔGSolv represents the variation between the GBSA solvation energy of the receptor–ligand complexes and the sum of the solvation energies for the protein and ligand. ΔGSA contains some of the surface area energies in the protein and ligand and the difference in the surface area energies for the complexes. The minimization of the docked complexes was done using a local optimization feature of prime.
MD Simulation
Desmond (Schrödinger Release 2018-4) by D.E. Shaw research was used to perform simulation of SARS-CoV2-PLpro in the presence of GRL0617 and ligand ZINC44459905 as it was having best docking score and able to mimic interactions of native ligand. To ensure the accuracy of results of MD simulation SARS-CoV2-PLpro-GRL0617 complex was used as a control, and results produced by SARS-CoV2-PLpro-ZINC44459905 complex were compared with it. For preparation of complexes and application of pre-simulation protein relaxation protein preparation wizard was used where the protein was prepared by performing restrained minimization using OPLS-2005 force field. The addition of hydrogens, water removal, bond orders assignment and fill in missing side chains and loops with optimization of hydrogen bond assignment (sampling of water orientations and use of pH 7.0) was done. LigPrep wizard of Maestro was used to prepare and minimize the ligands via addition of hydrogen atoms and generates tautomers, ionization states and ring conformations. Parameters for simulation were kept as following: solvent model: TIP3P; box shape: orthorhombic; dimension of box: 10 Å × 10 Å × 10 Å. Once these parameters were defined, Cl− or Na+ counter ions were used to perform neutralization. Steepest descent energy minimization was performed, and the simulation was proceeded for 100 ns with NPT (constant number of particles, pressure, and temperature) with 300 K (Thermostat method: Nose–Hoover chain) and 1.01 bar (Barostat method: Martyna–Tobias–Klein), constant volume, Smooth Particle Mesh Ewald (PME) method. After simulation, simulation interaction diagram wizard was used to analyze the trajectories for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), ligand–protein contact profiles and for ligand and protein modifications.
ADMET analysis
The ADMET properties of the GRL0617 and top five screened ligands were predicted using the pkCSM—pharmacokinetics server [29] that predicted both physiochemical and pharmacological properties. To perform this analysis SMILES (Simplified Molecule Input Line Entry Specification) of the compounds were retrieved from PubChem and uploaded to pkCSM—pharmacokinetics server. Parameters computed by server include water solubility in buffer system (SK atomic types, mg/L), in vivo CaCO2 cell permeability (human colorectal carcinoma), human intestinal absorption (HIA, %), in vivo P-glycoprotein inhibition and in vivo skin permeability (logKp, cm/hour). Metabolic parameters were determined using in vivo Cytochrome P450 2C19 inhibition, in vivo Cytochrome P450 2C9 inhibition, in vivo Cytochrome P450 2D6 inhibition, in vivo Cytochrome P450 2D6 substrate, in vivo Cytochrome P450 3A4 inhibition and in vivo Cytochrome P450 3A4 substrate. Distribution property included tests like blood–brain barrier (BBB) penetration, Lipinski’s rule (rule of five), central nervous system (CNS) permeability. To access the toxicity of compounds under study a range of important endpoints including acute algae toxicity, Ames test, 2-year carcinogenicity bioassay in mouse, 2-year carcinogenicity bioassay in rat, in vivo Ames test result in TA100 strain (metabolic activation by rat liver homogenate) were computed. Excretion again is a very important parameter and as many drugs often withdrawn at clinical trial stages due to their poorer renal clearance. In this study we included total renal clearance and renal OCT2 substrate to identify excretion efficacy of the proposed metabolite.
Results
Interactions produced by GRL0617 with SARS-CoV and SARS-CoV2-PLpro’s
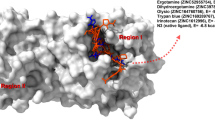
Multiple sequence alignment with ClustalW reveals 83% similarity with PLpro of SARS-CoV (3E9S) and SARS-CoV2 (7CMD). Furthermore, alignment reveals the presence of Tyr at position 269 in 3E9S and at 268 in 7CMD. Both proteins 3E9S and 7CMD were found to be superimposable and structurally identical also, and the pose of native co-crystallized ligand GRL0617 in binding cavity was also identical and superimposable. Side chain of Asp165 which is highly conserved amino acid among ubiquitin-specific protease (USP) family of deubiquitinating enzymes, forms hydrogen bonds with the amide group of GRL0617. Similarly, nitrogen present in backbone of Gln270 also participates in hydrogen bond formation. Apart from hydrogen bonds GRL0617 actively forms hydrophobic interaction with SARS-CoV-PLpro. In these interactions 1-napthyl group remains partly solvent exposed and interacts with four amino acids that are Tyr265 (Tyr264 for CoV2), Tyr269 (Tyr268 for CoV2), Pro248 (Pro247 for CoV2) and Pro249 (Pro248 for CoV2). Aromatic rings of Tyr and side chains of Pro are the active participants for these interactions. All these residues line the binding pocket and accommodate the leucine at the P4 position of PLpro substrates. Polar binding cavity of protein accommodates the (R)-methyl group, present at the stereocenter of the GRL0617, that points directly into the interior of the protein betweenTyr265 (Tyr264 for CoV2) and Thr302 (Thr301 for CoV2). On the other hand, R3 position of GRL0617 is occupied by –NH2 which extends from the opening cleft where series of polar groups such as the side chain oxygens of Gln270 (Gln269 for CoV2) and the hydroxyl of Tyr269 (Tyr268 for CoV2) could serve as a hydrogen bond accepter. Figures 2 and 3, respectively, represent the interaction of GRL0617 with PLpro of SARS-CoV and SARS-CoV2. Best docked ligand ZINC44459905 is having two rings in its structure, namely benzofuran and indole, that are linked together with -CONH bond. Both rings actively participate in formation of essential interactions with both proteins 3E9S and 7CMD. In 7CMD, benzofuran rings interact with four amino acids of binding cavity that are Glu167, Gly163, Tyr268 and Asp164. Here, Glu167 and Gly163, respectively, interact with benzene and furan rings of benzofuran.

Interaction profile of GRL0617 and best docked five antibacterial compounds from ZINC database with SARS-CoV-PLpro (PDB: 3E9S)

Interaction profile of GRL0617 and best docked five antibacterial compounds from ZINC database with SARS-CoV2-PLpro (PDB: 7CMD)
Docking antibacterial drugs with PLpro
Five drugs with ZINC id ZINC44459905, ZINC299856414, ZINC4639531, ZINC3896065 and ZINC24666638 produced significant binding energies along with multiple conserved interactions with Tyr269 (SARS-CoV-PLpro) and Tyr268 (SARS-CoV2-PLpro). Each of these drugs produced the binding energies of − 5.7, − 6.2, − 6.8, − 5.9 and − 8.1 kcal/mol for SARS-CoV-PLpro (3E9S), respectively, whereas for SARS-CoV2, binding energies were found to be − 7.3, − 7.0, − 7.2, − 7.1 and − 7.4 kcal/mol, respectively. Figures 2 and 3 represent the interactions produced by these drugs with both proteins. GRL0617 forms two conventional hydrogen bonds using hydrogen and oxygen of –NHCO, respectively, with Asp165 and Gln270 present in 3E9S, whereas in 7CMD three conventional hydrogen bonds are observed where two are formed in similar manner to 3E9S with Gln269 and Asp165.
One extra conventional hydrogen bond formation occurs between Gln269 and hydrogen of –NH2 present in benzamide ring. Furthermore, Asp of both proteins is also involved in formation of pi-anion bond with benzene ring of inhibitor. Apart from these bonds conserved amino acid Tyr268 in 7CMD and Tyr269 in 3E9S produces four different interactions. Tyr of both proteins forms single carbon hydrogen bond with –NHCO of GRL0617. Moreover, three hydrophobic T-shaped Pi-Pi interactions with naphthalene and benzene ring are also produced by Tyr of both proteins. Along with these interactions five different amino acids in both proteins (7CMD: Pro248, Pro247, Tyr273, Tyr264, Leu162; 3E9S: Pro249, Pro248, Tyr274, Tyr265, Leu163) also participate in formation of alkyl and pi-alkyl bonds. ZINC44459905, ligand with highest docking energy, mimicked the interactions produced by GRL0617. ZINC44459905 possesses two rings benzofuran and indole that are linked with –CONH bond. Benzene ring of benzofuran forms pi-anion interaction with side chain oxygen of Glu167 (7CMD) and Glu168 (3E9S), whereas furan ring interacts with three amino acids, namely: Asp, Gly and Tyr. Here, carbon of carboxy terminal of Gly163 (7CMD) and Gly164(3E9S) forms single pi-pi T-shaped interaction with furan ring. Asp164 (7CMD) forms three interactions where one pi-anion interaction is formed between furan ring and side chain oxygen of Asp, whereas other two interactions occur in form of carbon and conventional hydrogen bonds. Here, same side chain oxygen forms conventional hydrogen bond with hydrogen of –NH2 and carbon hydrogen bond with hydrogen of –CH2 linker that links indole ring and –CONH2-linked benzofuran ring. Similarly, Asp165 (3E9S) also produces the interactions with ligand in similar manner. Furthermore, Tyr264 of 7CMD produces alkyl and pi-alkyl interactions with benzamide and naphthalene ring linker in GRL0617 which is also produced by corresponding Tyr265 amino acid of 3E9S. In 7CMD docked with ZINC44459905 Tyr264 produces pi-sigma interaction rather than alkyl and pi-alkyl interactions, whereas corresponding Tyr265 of 3E9S does not participate in formation of any interaction with ZINC44459905. Hydrogen of N-terminal –NH2 of Gln269 (7CMD) and Gln270(3E9S) forms conventional hydrogen bond with oxygen of –CONH linker. Moreover, in 7CMD oxygen of delta carbon of sidechain of Gln269 forms conventional hydrogen bond with hydrogen of -NH2 of benzamide ring of GRL0617 which is absent on both ZINC44459905-3E9S and ZINC44459905-7CMD complex. Alkyl and pi-alkyl interactions between Pro249 (3E9S) (Pro248; 7CMD) and indole ring of ZINC44459905 are also mimicked. However, interactions of Pro248 (3E9S) (Pro247; 7CMD) are missing in both proteins with ZINC44459905 ligand. At last conserved amino acid Tyr268 (7CMD) and Tyr269(3E9S) mimic all four (three pi interactions and single hydrogen bond) interactions produced with native ligand which further represents the ZINC44459905 as strong analog of GRL0617. Apart from ZINC44459905, other ligands were also mimicking interactions of GRL0617, but their interaction with Tyr268 (7CMD) and binding energies was relatively weaker, and the type of interactions produced was also different than native ligand. Structural and chemical properties of GRL0617 along with screened drugs are given in Table 1 [30, 31].
MM-GBSA analysis
Receptor–ligand interaction causes modulation of energies of both free receptor and ligand (ΔGbind). Furthermore, these energies also have a significant impact on stability of receptor–ligand complex. In general, negative energies represent the higher stability of any system. Various energies produced during MM-GBSA for both 3E9S and 7CMD with native ligand GRL0617 as well as top five docking hits are presented in Table 2. Here, binding energies of GRL0617 with both 7CMD (− 67.5 kcal/mol) and 3E9S (− 73.1 kcal/mol) are in negative which suggests the higher spontaneity of interaction. Similarly, among tested compounds ZINC299856414 ranks highest in terms of stability (7CMD: − 55.67 kcal/mol), but the interaction with amino acid was found to be relatively weaker, whereas ZINC44459905 (7CMD: − 50.9 kcal/mol) was the second highest ranking compound that was producing stronger interactions with Tyr268. Binding energies and MM-GBSA energies of the rest of the tested compounds were also in negative, but the values were relatively higher which suggest low occurrence rate for the complex. Compound ZINC24666638 produced the highest docking score (7CMD: − 8.1 kcal/mol; 3E9S: − 7.4 kcal/mol) with PLpro of both viruses, but the ΔGbind energies were found to be the least negative (7CMD: − 21.08 kcal/mol; 3E9S: − 26.5 kcal/mol). Apart from ΔGbind energy, calculations for energy, hydrogen-bonding correction, lipophilic energy, Pi-pi packing correction and Van der Waals energy are also provided in Table 2.
Molecular dynamic simulations
ZINC44459905 was screened to be the best hit among tested compounds as it was producing consistent results in all parameters such as docking score, interactions and spontaneity. To validate these results further MD simulation of 100 ns was performed. Here, along with test compound (SARS-CoV2-PLpro-ZINC44459905), simulation of GRL0617 with SARS-CoV2-PLpro was also performed as a control set.
Once MD simulation was performed, the root-mean-square deviation (RMSD) values for all the frames present in trajectory were calculated. In general, RMSD value of any simulation represents the modulations that take place in the state of specific atoms with reference to their initial state. Here, to generate the RMSD value docked pose of protein–ligand complex is considered as an initial state or reference pose. Y-axis (left) of Fig. 4 represents the RMSD value of protein atoms. Analysis of this plot for SARS-CoV2-PLpro-GRL0617 (Fig. 4a) reveals that protein has equilibrated at the RMSD value of 2.0 ± 0.5 Å.

MD simulation protein–ligand interaction root-mean-square deviation (RMSD) profile of a SARS-CoV2-PLpro-GRL0617, b SARS-CoV2-PLpro- ZINC44459905
Here, the RMSD value is not exceeding 2.5 Å at the peak end which is good as for the smaller and most globular proteins RMSD value is ideally supposed to be in the range of 1–4 Å. For proteins that are bigger in size this value might exceed the upper boundary limit of 4 Å. For the complex of SARS-CoV2-PLpro-ZINC44459905 (Fig. 4b), RMSD value remains within the accepted limits. Here, similar to GRL0617 RMSD value is centered in the range of 2.0 ± 0.5 Å, but at the end of the simulation RMSD value of protein increases and reaches up to 2.7 Å and immediately after that fall back into the normal range which suggests temporary shift in the protein structure. In the graphs of Fig. 4 right Y-axis represents the ligand RMSD value which provides the insight on the stability of the docked ligand pose in the binding pocket. ‘Lig Fit Prot’ signifies the RMSD values of ligand with reference to protein backbone. This value is supposed to be slightly higher than that of protein RMSD value, but the presence of significantly higher value suggests the major changes in ligand pose compared to that of docked pose. Throughout the whole simulation Lig Fit Prot value of SARS-CoV2-PLpro-GRL0617 (Fig. 4a) complex is in the range of 2.5 ± 1 Å, whereas, for the SARS-CoV2-PLpro-ZINC44459905 complex, this value is within the range of 2.6 ± 1 Å, but similar to protein RMSD value, ligand RMSD value also shows the temporary shift at the end of simulation. On the other hand, Lig Fit Lig value of GRL0617 is lower than that of the ZINC44459905, but the difference is of only 0.4 Å which is acceptable. Such results show that GRL0617 is having relatively higher stability compared to the tested ligand in binding pocket at the given docking pose.
To study the smaller changes across the entire protein during simulation the root-mean-square fluctuation (RMSF) plots of both native and tested ligand were studied (Fig. 5). Here, left Y-axis represents the protein RMSF values, whereas right Y-axis shows the experimental B-factor values that are computed during the process of crystal structure refinement. In general, RMSF values and B-factor values of protein are supposed to be parallel, but as the fundamental ideas of both are different, one-to-one correspondence is not possible. In the current study, RMSF and B-factor values were found to be parallel which demonstrates the reliability of produced data. Sharp peaks in the plots represent the regions of protein that are varying constantly and frequently throughout the simulation. In general, tail regions of any protein are least stable compared to the internal regions; hence, highest fluctuation is observed in these regions. Among internal regions alpha helix and beta-sheets have the least flexibility; hence, these regions of protein remain constant for more than 70% of the simulation period. The most fluctuating and dynamic regions are the unstructured regions. In the plot, regions of protein that interacts with ligand are depicted with green vertical bars. Here, all the motions observed in the structured and unstructured regions of test ligand–protein complex were similar to that of control ligand–protein complex.

MD simulation protein–ligand interaction root-mean-square fluctuation (RMSF) profile of a SARS-CoV2-PLpro-GRL0617, b SARS-CoV2-PLpro- ZINC44459905
For the extensive study of the interactions produced during the course of simulation three different plots presented in Figs. 6, 7 and 8 were used. Hydrogen bonds, hydrophobic interactions such as Pi cation, Pi-Pi stacking, water bridges and ionic interactions were monitored. Among these variety of interactions ionic interactions were found to be absent in both control and test ligand. Hydrogen bonds are very much crucial in determining the specificity, metabolization and adsorption of drugs. Hence, for the complex of SARS-CoV2-PLpro-GRL0617 hydrogen bonds were observed. Five different amino acids: Asp164, Tyr264, Tyr268, Gln269 and Tyr273 formulated the hydrogen bonds with ligand for varying periods. Among these hydrogen bonds three Tyr264, 268 and 273 were observed for less than 10% of the time, whereas Asp164 was forming this interaction for 26% of the time. Most stable hydrogen bond was formulated by Gln269 with oxygen of the –CONH, which was noted for the 99% of the time. Furthermore, amino acids that participated in hydrogen bond formation also formulated the water bridges except for Tyr264. Also, Glu167 was found to be participating in water bridge formation for 10% of the time; this particular interaction was found to be frequently occurring during the end phase of the simulation. Water bridges are relaxed form of hydrogen bonds that are being formulated between protein and ligand via intermediatory water molecules. For SARS-CoV2- ZINC44459905 complex, direct hydrogen bonds were formulated by all the amino acids; similar to GRL0617 additional two weak bonds were observed with ARG166 and Thr301 for a very brief period of time. Gln269 produced the hydrogen bond with highest stability of 60% of total time. All the amino acids that were forming water bridges with GRL0617 were also producing the water bridges with ZINC44459905 except for the Glu167. In addition, Arg166, Ala246, Tyr264, Asn267, Cys270, Gly271, Thr301 and Asp302 were also able to produce water bridges. Apart from hydrogen bonds five different amino acids Leu162, Pro247, Pro248, Tyr264, Tyr268 and Tyr273 were participating in formation of hydrophobic interactions. Here, Tyr273 and Tyr247 produced the hydrophobic interaction for less than 20% of total time, whereas Leu162 produced the interaction for less than 40% of time. Pro248 and Tyr264 produced the interaction for nearly 60% of the time, whereas highest hydrophobic interactions were maintained by Tyr268. For SARS-CoV2-ZINC44459905 complex, all the same amino acids were forming hydrophobic interactions. Additionally, Lys157 was found to be participating in such interactions. Analysis of the all the graphs and plots produced during MD simulation confirms the findings of docking.

Protein–ligand interaction profile of SARS-CoV2-PLpro-GRL0617 complex a interaction profile of crucial interacting amino acids, b ligand interaction diagram showing percent of total time a particular interaction is involved

Protein–ligand interaction profile of SARS-CoV2-PLpro-ZINC44459905 complex a interaction profile of crucial interacting amino acids, b ligand interaction diagram showing percent of total time a particular interaction is involved

Timeline representation of the interactions of ligand with amino acids for the complex a SARS-CoV2-PLpro-GRL0617, b SARS-CoV2-PLpro-ZINC44459905
ADMET analysis
All the ADMET properties of the test compounds along with control compound GRL0617 are presented in Table 3. For any drugs to work on human body five properties play very crucial role that are absorption, distribution, metabolism, excretion and toxicity. Property of absorption was measured using seven different models. Here, to assess the effectiveness of selected compound as oral drug CaCO2 permeability and intestinal absorption were measured. Analysis revealed that all the compounds except ZINC4639531 and ZINC24666638 have higher values for CaCO2 permeability. Furthermore, both compounds were also having lowest intestinal absorption, whereas ZINC44459905 is having 77% absorption. Moreover, assessment of skin permeability was lowest for all the tested as well as control compounds which suggest minimum absorption. To evaluate the transport of the compounds across the membrane three tests for P-glycoprotein were performed where test compounds were tested as a substrate of the P-glycoprotein and inhibitors of the P-glycoprotein I and II. All the compounds were found to be the compatible substrates which suggests that they can pass across the membrane using ATP-binding cassette (ABC) transporter, whereas assay as an inhibitor of P-glycoprotein I and II was found to be negative for ZINC44459905 which highlights the inability of this compound to inhibit both these efflux pumps of foreign substances. To measure the distribution of the compounds throughout the body four assays were used, namely they are volume of distribution (VDss), fraction unbound, blood–brain barrier (BBB) permeability and central nervous system (CNS) permeability. VDss assay is used to measure the total quantity of drugs needed for uniform distribution of drugs throughout the blood. Values for the assay for all the compounds were found to be lower than 0.45 LogVDss which suggests that lower drug volume will be required. For ZINC44459905 this value was found to be the least − 1.056 LogVDss. BBB permeability of the ZINC44459905 was found to be − 0.646 which suggests lower permeability; similarly, values for CNS permeability were also − 2.409. Hence, compound is predicted to have lower permeability for CNS. Metabolism of the test drugs within body was evaluated using seven different models of cytochrome where test compounds were evaluated for their ability to act as a substrate for CYP2D6 and CYP3A4. Further these compounds were assayed for the inhibition of CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4. ZINC44459905 was found to be negative for all the categories except for the CYP2C9 inhibition. Excretion of the compounds from the body was assessed using two models that are total clearance and renal OCT2 assay. All the test as well as control compounds were found to be negative for the renal OCT2 assay; hence, none of them can be excreted using organic cation transporter 2, whereas total clearance assessment produces the value of 0.595 mL/min/kg for ZINC44459905 which is higher than GRL0617. Toxicity assessment was carried out with 10 different models. ZINC44459905 and ZINC24666638 were found to be negative for the AMES test which suggests that these compounds are not carcinogenic or mutagenic, whereas rest of the compounds including GRL0617 were reported positive for this test. For the ZINC44459905 maximum recommended tolerated dose value was found to be highest that is 1.119 log(mg/kg/day). Among the drugs hERG (human ether-a-go-go gene) I and II inhibitors are not favored as inhibition of this genes has been reported to cause QT syndrome which can lead to the fatal ventricular arrhythmia. Here, all the drugs including control were found to be negative as the inhibitor of hERG I, whereas for hERG II, two compounds ZINC44459905 and ZINC24666638 were found to be negative as an inhibitor. Oral rat acute and chronic toxicity values for ZINC44459905 were higher than that of GRL0617 which suggests that relatively higher doses of the compound are producing toxicity. Detailed values of the ADMET analysis are given in Table 3.
Discussion
COVID consists of group of viruses known to infect humans and other animals. Biological features of this group of viruses mostly remain the same such as genomic material consists of positive strand of RNA, and S-, E-, M- and N-proteins are the most common protein found in all the members of the group. These viruses belong to subfamily coronavirinae which is further separated into four different genus that are namely: (I) alpha coronavirus, (ii) beta coronavirus, (iii) gamma coronavirus, (vii) delta coronavirus. Here, alpha and beta coronaviruses are frequently observed in humans and animals, whereas gamma and delta primarily target the birds. For instance, MERS-CoV and SARS-CoV both are the beta coronavirus that, respectively, causes Middle East Respiratory Syndrome, or MERS and Serious Intense Respiratory Disorder, or SARS. Our most recent opponent SARS-CoV-2, causative agent of COVID-19, is also the member of beta coronaviruses. Apart from these viruses, Porcine Transmissible Gastroenteritis Virus (TGEV), Bovine Coronavirus (BCV), Avian Infectious Bronchitis Virus (IBV), Feline Infectious Peritonitis Virus (FIPV), Canine Coronavirus (CCoV), Porcine Hemagglutinating Encephalomyelitis Virus (HEV) and Turkey Coronavirus are also members of coronavirinae subfamily [32], but they are unable to infect humans as they lack the necessary S-proteins for cellular entry. MERS and SARS- CoV have S-protein that can interact with ACE2 receptor and facilitate the viral entry in human cells [33,34,35]. S-proteins are specific for hosts; hence, cross-infection is uncommon, but on occasions it has been noted, for instance SARS-CoV (2003) was transferred to humans from bats. Similarly, MERS-CoV (2005) was primarily infecting camels, but cross-infections in humans were also noted. Recently, member of COVID group, SARS-CoV2, is noted as infectious agents in humans due to cross-infectivity. Moreover, such instances are frequently occurring; hence, it is not farfetched to assume that it will not be the last instance of such nature [8, 36].
Apart from vaccine development, scientific communities have also devoted their energies and resources to identify compounds that can counter the SARS-CoV2 virus. For identification of such compounds docking, MD simulations, pharmacophore and ADMET profiling are frequently utilized methods [37]. Variety of natural products from bacteria, fungi and plants have been explored to inhibit the necessary proteins of virus as their natural origin makes them vastly available. Moreover, instances of toxicity are also limited for such compounds. For instance, pyranonigrin A and flaviolin are the compounds produced by fungi. Their docking and simulation studies with Mpro suggest that they can inhibit the viral replication via interacting with this protein [5]. Drug repurposing is another approach that has been utilized by number of researchers where existing, commercial drugs have been pitted against viral proteins with hope that one of them might produce good result. Procainamide, tetrahydrozoline, levamisole are the drugs that have been found to be effective against the papain-like protease of SARS-CoV-2 [38]. All the above-mentioned studies have been carried out in silico using molecular dynamic simulation and docking studies. In addition, these approaches have also been utilized for potency assessment of hydroxychloroquine against various potential targets of SARS-CoV2 [39]. In all of these studies the rationale is very basic that if any of these drugs can meddle with the viral proteins and targets, then they can hinder the normal life cycle of the virus [40, 41].
With the concept of drug repurposing several attempts have been made to identify the potential inhibitors of PLpro [12, 42]. Among library of 3727 approved drugs, remdesivir and hydroxychloroquine have been reported as potential inhibitors of PLpro as they have been found to be interacting with it. Furthermore, results of docking and MD were validated using in vitro assays where effective results were produced by both proposed compounds [12]. IBS database (https://www.ibscreen.com/natural.shtml) library of 50,000 natural compounds has been docked against the PLpro where the compounds with best dock score were STOCK1N-69160 [(S)- 2-((R)-4-((R)-2-amino-3-methylbutanamido)-3-(4-chlorophenyl)butanamido)propanoic acid hydrochloride] and STOCK1N-69160 [(S)-2-((R)-4-((R)-2-amino-3- methylbutanamido)-3-(4-chloro- phenyl) butanamido)propanoic acid hydrochloride]. Regardless, these compounds were found to be making single hydrophobic pi-pi stacking interaction with Tyr268 where native ligand GRL0617 produces three hydrophobic and one hydrogen bond interaction; moreover, native ligand GRL0617 or any other known inhibitor of PLpro was not utilized as a control for the entire study [43]. In the present study, repurposed antibacterial drugs managed to produce significantly better interactions with Tyr268 and GRL0617 were utilized as a positive control. Among various compounds that have been identified as an inhibitor of PLpro GRL0617 has been recognized as the most effective inhibitor of PLpro which further increases the reliability of the produced results in current in-silico study [42]. In vitro validation of the proposed compounds necessitates the utilization of Biosafety Level 4 (BSL4); as we are dealing with infecting agents, only in-silico study has been carried out. Here, it is hoped that the people with such facility can validate these in-silico results. There are several studies with in-silico reports, but there are few proving the findings with in vitro anti-viral assays. In one such article by Jamalan and colleagues [44], a molecule 5-(aminomethyl)-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl]-benzamide (ZINC43071312) was shown to effectively bind with PLpro and showed SARS-CoV-PLpro activity with an IC50 of 460 nM; moreover, for SARS-CoV2-PLpro, this molecule was found to interact with Tyr268, Pro248, Thr301, Pro247, Tyr273 and Tyr264; in comparison, the top hit of our study ZINC44459905 showed to interact with Gly163, Asp164, Glu167, Pro248, Tyr264, Tyr268, Gln269, thus showing the compound under study recruits amino acids that are essential to induce inhibition of PLpro.
Here, in the current study 8581 compounds that have been reported active against bacteria were downloaded from the ZINC database and they were manually filtered to remove the compounds that were too large. Compounds with 250–400 molecular weight were separated and utilized for the docking. GRL0617 is having one naphthalene moiety in the structure which participates in formation of hydrophobic pi interactions and hydrogen bond with Tyr268. Hence, compounds with rings to form pi interaction and central –CONH to from hydrogen bond with Tyr268 were prioritized [6]. Here, five top hits have been proposed as an antibacterial drug that targets different proteins that are exclusive to bacteria. ZINC44459905 best docked compound has been utilized as an inhibitor of biotin carboxylase enzyme [45]. In view of unavailability of medication for corona virus infection, the present study proposes ZINC44459905 as the lead molecule that interact with Tyr268 of SARS-CoV2-PLpro to inhibit its function and can be beneficial in forthcoming in vitro and in vivo studies for COVID-19 therapeutics.
References
Chen Y, Guo Y, Pan Y, Zhao ZJ (2020) Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2020.02.071
Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, Zhu H, Zhao W, Han Y, Qin C (2019) From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. https://doi.org/10.3390/v11010059
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YZYZL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZYZL (2020) A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. https://doi.org/10.1038/s41586-020-2008-3
Schwartz DA, Graham AL (2020) Potential maternal and infant outcomes from coronavirus 2019-NCOV (SARS-CoV-2) infecting pregnant women: lessons from SARS MERS, and other human coronavirus infections. Viruses. https://doi.org/10.3390/v12020194
Rao P, Shukla A, Parmar P, Rawal RM, Patel BV, Saraf M, Goswami D (2020) Proposing a fungal metabolite-flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using docking and molecular dynamics. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1813202
Rao P, Patel R, Shukla A, Parmar P, Rawal RM, Saraf M, Goswami D (2021) Identifying structural-functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol Divers. https://doi.org/10.1007/s11030-021-10220-8
Parmar P, Rao P, Sharma A, Shukla A, Rawal RM, Saraf M, Patel BV, Goswami D (2021) Meticulous assessment of natural compounds from NPASS database for identifying analogue of GRL0617, the only known inhibitor for SARS-CoV2 papain-like protease (PLpro) using rigorous computational workflow. Mol Divers. https://doi.org/10.1007/s11030-021-10233-3
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Kong R, Yang G, Xue R, Liu M, Wang F, Hu J, Guo X, Chang S (2020) COVID-19 Docking server: an interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19, J ArXiv Prepr
Kong FD, Yi TF, Ma QY, Xie QY, Zhou LM, Chen JP, Dai HF, Wu YG, Zhao YX (2020) Biphenyl metabolites from the patchouli endophytic fungus alternaria sp PfuH1. Fitoterapia 146:104708. https://doi.org/10.1016/j.fitote.2020.104708
McClain CB, Vabret N (2020) SARS-CoV-2: the many pros of targeting PLpro. Signal Transduct Target Ther 5:1–2. https://doi.org/10.1038/s41392-020-00335-z
Klemm T, Ebert G, Calleja DJ, Allison CC, Richardson LW, Bernardini JP, Lu BG, Kuchel NW, Grohmann C, Shibata Y, Gan ZY, Cooney JP, Doerflinger M, Au AE, Blackmore TR, van Heden GJN, Geurink PP, Ovaa H, Newman J, Riboldi-Tunnicliffe A, Czabotar PE, Mitchell JP, Feltham R, Lechtenberg BC, Lowes KN, Dewson G, Pellegrini M, Lessene G, Komander D (2020) Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 39:1–17. https://doi.org/10.15252/embj.2020106275
Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden GJ, van Noort H, Ovaa S, Müller KP, Knobeloch K, Rajalingam BA, Schulman J, Cinatl G, Hummer S, Ciesek ID (2020) Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587:657–662. https://doi.org/10.1038/s41586-020-2601-5
Pushpakom S, Iorio F, Eyers PA, Jane Escott K, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, Norris A, Sanseau P, Cavalla D, Pirmohamed M (2018) Drug repurposing: progress, challenges and recommendations. Lancet Oncol. https://doi.org/10.1038/nrd.2018.168
Muratov EN, Amaro R, Andrade CH, Brown N, Ekins S, Fourches D, Isayev O, Kozakov D, Medina-Franco JL, Merz KM, Oprea TI, Poroikov V, Schneider G, Todd MH, Varnek A, Winkler DA, Zakharov AV, Cherkasov A, Tropsha A (2021) A critical overview of computational approaches employed for COVID-19 drug discovery. Chem Soc Rev. https://doi.org/10.1039/D0CS01065K
Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Sastry GM, Adzhigirey M, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the. J Chem Theor Comput 6:1509–1519
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J Med Chem 47:1750–1759. https://doi.org/10.1021/jm030644s
Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C, Li Y, Bai F, Wang H, Cheng X, Cen X, Hu S, Yang X, Wang J, Liu X, Xiao G, Jiang H, Rao Z, Zhang LK, Xu Y, Yang H, Liu H (2020) Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (80-) 368:1331–1335. https://doi.org/10.1126/science.abb4489
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691. https://doi.org/10.1007/s10822-007-9133-z
Trott O, Olson AJ (2010) AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem 49:6177–6196. https://doi.org/10.1021/jm051256o
Halgren T (2007) New method for fast and accurate binding-site identification and analysis. Chem Biol Drug Des 69:146–148. https://doi.org/10.1111/j.1747-0285.2007.00483.x
Wang W, Donini O, Reyes CM, Kollman PA (2001) Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct 30:211–243. https://doi.org/10.1146/annurev.biophys.30.1.211
Wang J, Hou T, Xu X (2006) Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr Comput Aided-Drug Des 2:287–306. https://doi.org/10.2174/157340906778226454
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897. https://doi.org/10.1021/ar000033j
Massova I, Kollman PA (2000) Combined molecular mechanical and continuum solvent approach (MM- PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des 18:113–135. https://doi.org/10.1023/A:1008763014207
Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
López-López E, Naveja JJ, Medina-Franco JL (2019) DataWarrior: an evaluation of the open-source drug discovery tool. Expert Opin Drug Discov 14:335–341. https://doi.org/10.1080/17460441.2019.1581170
Sander T, Freyss J, von Korff M, Rufener C (2015) DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model 55:460–473. https://doi.org/10.1021/CI500588J
Shukla A, Parmar P, Kapoor G, Goswami D, Jha CK, Patel B, Saraf M (2021) Curse of La corona: unravelling the scientific and psychological conundrums of the 21st century pandemic. Mol Divers. https://doi.org/10.1007/s11030-020-10167-2
Ren Z, Yan L, Zhang N, Guo Y, Yang C, Lou Z, Rao Z (2013) The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles’’ heel": current effective inhibitor targeting a 3C-like protease”. Protein Cell 4:248–250. https://doi.org/10.1007/s13238-013-2841-3
Kumar A, Mehta V, Raj U, Varadwaj PK, Udayabanu M, Yennamalli RM, Singh TR (2018) Computational and in-vitro validation of natural molecules as potential acetylcholinesterase inhibitors and neuroprotective agents. Curr Alzheimer Res 16:116–127. https://doi.org/10.2174/1567205016666181212155147
Yang H, Bartlam M, Rao Z (2006) Drug design targeting the main protease, the achilles heel of coronaviruses. Curr Pharm Des 12:4573–4590. https://doi.org/10.2174/138161206779010369
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. https://doi.org/10.1038/s41586-020-2012-7
Santibáñez-Morán MG, López-López E, Prieto-Martínez FD, Sánchez-Cruz N, Medina-Franco JL (2020) Consensus virtual screening of dark chemical matter and food chemicals uncover potential inhibitors of SARS-CoV-2 main protease. RSC Adv 10:25089–25099. https://doi.org/10.1039/D0RA04922K
Arya R, Das A, Prashar V, Kumar (2020), Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs
Mukherjee S, Dasgupta S, Adhikary T, Adhikari U, Panja SS (2020) Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1804458
Parmar P, Shukla A, Rao P, Saraf M, Patel B, Goswami D (2020) The rise of gingerol as anti-QS molecule: darkest episode in the LuxR-mediated bioluminescence saga. Bioorg Chem 99:103823. https://doi.org/10.1016/j.bioorg.2020.103823
Pandya PN, Kumar SP, Bhadresha K, Patel CN, Patel SK, Rawal RM, Mankad AU (2020) Identification of promising compounds from curry tree with cyclooxygenase inhibitory potential using a combination of machine learning, molecular docking, dynamics simulations and binding free energy calculations. Mol Simul 46:812–822. https://doi.org/10.1080/08927022.2020.1764552
France A, Hognon C, Miclot T, García-Iriepa C, Iriepa I, Terenzi A, Grandemange P, Barone G, Marazzi M, Monari A (2020) Molecular basis of SARS-CoV-2 infection and rational design of potential antiviral agents: modeling and simulation approaches. Cite This J Proteome Res 19:4291–4315. https://doi.org/10.1021/acs.jproteome.0c00779
Amin SA, Ghosh K, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: monte carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1780946
Jamalan M, Barzegari E, Gholami-Borujeni F (2020) Structure-based screening to discover new inhibitors for papain-like proteinase of SARS-CoV-2: an in silico study. J Proteome Res 20:1015–1026. https://doi.org/10.1021/ACS.JPROTEOME.0C00836
Škedelj V, Tomašić T, Mašič LP, Zega A (2011) ATP-binding site of bacterial enzymes as a target for antibacterial drug design. J Med Chem 54:915–929. https://doi.org/10.1021/jm101121s
Acknowledgements
Authors are thankful to Department of Microbiology and Biotechnology, School of Sciences, Gujarat University, DST-FIST Sponsored Department, for providing necessary facilities to perform experiments. Authors are also thankful to University Grants Commission (UGC), New Delhi, India, for providing the fellowship for the award of ‘CSIR-NET Junior Research Fellowship (JRF)’ to Jignesh Prajapati. Authors, Rohit Patel and Priyashi Rao, are thankful to ScHeme of Developing High quality research (SHODH), Education department, Government of Gujarat, India, for providing fellowship. We acknowledge GSBTM, DST, Government of Gujarat, for providing Bioinformatics Node facility and Finishing School support. We acknowledge GUJCOST, DST, Government of Gujarat, for Super-computing facility provision.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Patel, R., Prajapati, J., Rao, P. et al. Repurposing the antibacterial drugs for inhibition of SARS-CoV2-PLpro using molecular docking, MD simulation and binding energy calculation. Mol Divers 26, 2189–2209 (2022). https://doi.org/10.1007/s11030-021-10325-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10325-0