
Abstract
The outbreak of SARS-CoV-2 and deaths caused by it all over the world have imposed great concern on the scientific community to develop potential drugs to combat Coronavirus disease-19 (COVID-19). In this regard, lichen metabolites may offer a vast reservoir for the discovery of antiviral drug candidates. Therefore, to find novel compounds against COVID-19, we created a library of 412 lichen compounds and subjected to virtual screening against the SARS-CoV-2 Main protease (Mpro). All the ligands were virtually screened, and 27 compounds were found to have high affinity with Mpro. These compounds were assessed for drug-likeness analysis where two compounds were found to fit well for redocking studies. Molecular docking, drug-likeness, X-Score, and toxicity analysis resulting in two lichen compounds, Calycin and Rhizocarpic acid with Mpro-inhibiting activity. These compounds were finally subjected to molecular dynamics simulation to compare the dynamics behavior and stability of the Mpro after ligand binding. The binding energy was calculated by MM-PBSA method to determine the intermolecular protein–ligand interactions. Our results showed that two compounds; Calycin and Rhizocarpic acid had the binding free energy of − 42.42 kJ mol/1 and − 57.85 kJ mol/1 respectively as compared to reference X77 (− 91.78 kJ mol/1). We concluded that Calycin and Rhizocarpic acid show considerable structural and pharmacological properties and they can be used as hit compounds to develop potential antiviral agents against SARS-CoV-2. These lichen compounds may be a suitable candidate for further experimental analysis.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recently, Novel Coronavirus (SARS-CoV-2) is spreading very rapidly all over the world and causing an ongoing outbreak of COVID-19, a serious and often fatal respiratory tract infection. COVID-19 has created an emergency in India and all around the world. The SARS-CoV-2, previously named as 2019 novel coronavirus (2019-nCoV), is a positive-sense, single-strand RNA coronavirus. According to the Worldometer's report, about 81,285 people in China have been infected with coronavirus and 3287 deaths since its emergence in the city of Wuhan, Hubei province. After China, USA and Italy have been affected most from the coronavirus followed by Spain, Germany Iran, and France. The outbreak of coronavirus is increasing day by day. By now, more than 1,804,128 people have been diagnosed, and more than 112,223 deaths have been recorded worldwide from COVID-19, according to World Health Organization (WHO) figures till 13 April, 2020. The outbreak of corona is also affecting India and to date 13 April 2020, total 8447 total cases and 273 death reports from COVID-19.
Due to its disseminating rate and fatality, COVID-19 is declared as a pandemic disease by WHO to coordinate scientific and medical efforts to rapidly develop a cure for patients. Currently, there is no appropriate vaccines and antiviral agents are available that can effectively prevent or treat the COVID-19 infection, and mortality is increasing day by day. This situation is putting the whole world under high pressure to develop novel vaccines or drugs against it. On the date, March 17, 2020, the USA reported starting vaccine trial against COVID-19 but it will take more than one year to come in markets. Therefore, effective treatment or control mechanism is needed to prevent Coronavirus [1]. Consequently, to develop new drugs against COVID-19, we have conducted computational screening of compounds from lichen species which may be a natural treasure for many types of pharmacologically active compounds against coronavirus. Many lichen species have been reported to have antiviral, antibacterial, and antifungal activity, etc. [2, 3]. Various scientific reports have suggested that lichen metabolites may a valuable treasure for antiviral drug candidates [3,4,5]. Recently, in March 2020, a group of researchers at UBC screened 1.3 billion small molecules for potential inhibitors against the SARS-CoV-2 Main Protease by deep docking method [6]. Hence, we were curious whether lichen compounds can also prevent SARS-CoV-2 or not and carried out virtual screening to find out potential natural anti-SARS-CoV-2 agents.
For the control of viral replication, inhibition of replication of viral genomic material is a good strategy for potential antiviral drug discovery [7]. Given that SARS-CoV-2 is a (+) SS RNA virus, its main protease (Mpro) cut two replicase polyproteins, which is required to mediate viral replication and transcription, Mpro can be used as a molecular target for drug discovery. By inhibiting the Mpro protein, virus replication could be stopped. Therefore, we selected viral Mpro enzyme as a drug target to quickly identify novel inhibitors of SARS-CoV-2.
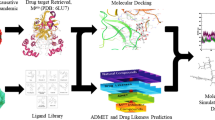
To achieve this aim, 412 lichen compounds were selected for molecular docking using AutoDock Vina, based on a literature search, to explore their binding modes with Mpro. Furthermore, all screened hits were subjected to a drug-likeness investigation based on physiochemical properties using DruLiTo tool. The screened compounds having drug-like property and high binding affinity with target protein were taken for rescoring using X-Score. Further, all screened hits were subjected to extensive toxicity analysis using the OSIRIS Property Explorer. Protein–ligand molecular interaction of compounds with remarkable inhibitory characteristics against the selected target protein was viewed with PyMOL and LigPlus to gain structural insight into the binding interaction, including the types of bonding interaction and the amino acids involved in such interactions, compared to its standard inhibitor. Finally all screened compounds were subjected to Molecular Dynamics Simulation (MDS) and Molecular mechanic/Poisson–Boltzmann Surface Area (MM-PBSA) analysis to understand the stability as well as dynamics of the protein–ligand complexes. The schematic representation of the methodology is shown in Fig. 1.

Schematic representation of various steps of the methodology
Materials and methods
Construction of library
Text mining analysis by using DLAD4U, PubTator, and Carrot2 servers showed that several lichen spp. have antiviral properties. Hence to screen antiviral compounds against coronavirus, a library of 412 Lichen compounds was built in-house by collecting information from scientific literature [8] and Herbal Net Digital Repository database [http://herbalnet.healthrepository.org/]. The compounds with known 2D structures in PubChem [https://pubchem.ncbi.nlm.nih.gov] and ChemSpider databases [http://www.chemspider.com] were considered to create the library [9]. The library of lichen compounds is shown in Supplementary Table S1.
Receptors preparation
The COVID-19 main protease (Mpro) was selected as the target protein in this study. The X-ray crystal structure of COVID-19 Mpro covalently attached with their non-peptide inhibitor D3F (PDB ID- 6W63) was retrieved from the Protein Data Bank [http://www.rcsb.org/pdb/home/home.do]. The water molecules and ions were removed from the protein molecule using PyMOL software. After that, the addition of hydrogen atoms to the receptor molecule was carried out by using AutoDockTools (ADT). Thereafter, nonpolar hydrogens were merged,while polar hydrogen were added to the protein. Subsequently, the protein was saved into a dockable pdbqt format for molecular docking analysis (Fig. 2a).

X-ray crystal structure of COVID-19 Mpro covalently linked to X77 (a), the superimposition of the docked X77 with its X-ray crystal structure. Blue and orange color indicates experimental and docked X77 respectively (b), and 2D interaction of experimental and docked X77 with Mpro (c). The green dotted lines and attached residues indicate H-bond and H-bonding residues, residues present in the half-circle represent the hydrophobic bond-forming residues and red circle shows the active site residues
Ligand preparation
The 3D structure of the reference molecule, X77 which was co-crystallized with Mpro was retrieved from the respective protein from Protein Data Bank. The 3D structure of each ligand (lichen compounds) was obtained from various online resources and compound databases, e.g., PubChem, ZINC, and CHEM SPIDER in MOL or SDF format. The compounds were converted to MOL2 chemical format using Open babel. Polar hydrogen charges were assigned and the nonpolar hydrogens were merged by using Autodock tools. Finally, the compounds were further converted to the dockable pdbqt format for molecular docking.
Molecular docking
To achieve the mode of interaction of Lichen compounds with the binding pocket of COVID-19 Mpro, molecular docking was performed by using AutoDock Vina software in PyRx platform (GUI version 0.8). Validation of docking protocol is done by performing the docking of the co-crystallized ligand at the active site of the 3D structure of the same protein. Therefore, docking was performed with reference molecule X77, and the RMSD value between experimental and docked reference was calculated to validate the docking protocol. RMSD has often been used to measure the quality of reproduction of a known binding pose by a computational method similar to the crystallized protein–ligand complex. Lower the value of RMSD reflects higher the accuracy of docking and RMSD values less than 2.0 Angstrom are significantly good to consider [10]. Using PyRx software, the grid box centered for Mpro was X = − 23.30, Y = 12.84, and Z = − 29.66 and the dimensions of the grid box were set as 66.93 × 37.59 × 31.68 Å. After validation of the docking protocol, virtual screening was conducted by rigid docking into the active site of protein. The binding modes were clustered through the root-mean-square deviation (RMSD) among the coordinates of the ligand atoms. The compounds were then ranked by their binding affinity scores. Thereafter, molecular interaction between Mpro and compounds with a binding affinity higher than that of the respective reference compound was viewed with PyMOL.
Drug-likeness analysis
The pharmacological significance of any compound is based on its drug-likeness which is calculated based on certain physicochemical properties. Therefore, all ligands were evaluated for its drug-like nature by DruLiTo software. Lipinski’s rule of five (RO5) which is considered as an empirical thumb rule was used to filter the compounds based on their drug-likeness property [11]. In addition to Lipinski RO5, PAINS filter was also used in drug-likeness analysis to filter the false positive from the screened hits by using PAINS remover [12].
Toxicity risk prediction
The compounds having drug-like property and good binding affinity with Mpro receptor were taken for the extensive toxicity analysis using the OSIRIS Property Explorer [13].
Rescoring of docking results
The best molecules from molecular docking, drug-likeness, and toxicity analysis were re-docked using the X-SCORE program [14]. X-Score is a scoring function to calculate the binding affinity of the ligand molecules toward their target protein. The same binding pocket was selected for docking studies that were used in virtual screening. Two different kinds of files required in X-Score is a receptor structure file (in PDB format) and a ligand structure file (in MOL2 format). Both the structure files were used as an input file to carry out X-score analysis. After that binding energy of the protein–ligand complex was calculated by using the X-score command. All default parameters of X-Score were used. The X-Score program uses three scoring functions, viz. HP Score, HM Score, and HS Score. The final X-Score = (HP Score + HM Score + HS Score).
Visualization
PyMOL was used to visualize the docked pose of screened compounds at the active site of the receptor. The 2D interactions of the protein–ligand complexes were performed by LigPlot + v.1.4.5 program to identify the interactions of an amino acid between protein and ligand. LigPlot depicted hydrophobic bonds, hydrogen bonds, and their bond lengths in each docked pose.
Molecular dynamic simulation
For predicting the stability of Mpro and Mpro-ligand complex, molecular dynamics simulations (MDS) were performed in a GROMACS 5.0 [15] package as per the protocol described by publication [9, 16]. The MD simulations were executed on a work station with configuration Ubuntu 16.04 LTS 64-bit, 4 GB RAM, Intel®CoreTM i5-6400 CPU. Three systems (two Mpro-screened ligand complex and a Mpro-reference ligand complex) were created and subjected for 10 ns MD simulation studies. The topology file for ligand and protein was generated by using CGenFF server and ‘pdb2gmx’ script, respectively, by using CHARMM 36 force field [17]. After that ligand topologies were rejoined to the processed protein structure for building the complex system. After that, a water solvated system was built by using the TIP3P water model with dodecahedral periodic boundary conditions. The solutes are centered in the simulation box with a minimum distance to the box edge of 10 Å (1.0 nm). After defining the box, all the systems were solvated using the TIP3P water model in a dodecahedral box and neutralized by adding Na + counter-ions by using the ‘gmx genion’ script. Energy minimization of the complexes was done at 10 kJ/mol with steepest descent Algorithm by using Verlet cut off-scheme taking Particle Mesh Edward (PME) Coulombic interactions with a maximum of 50,000 steps. The equilibration of the system was obtained in two steps. In the first step, NVT equilibration was done in 300 K and 5000 ps of steps, while in the second step, NPT equilibration taking Parrinello-Rahman (pressure coupling), 1 bar reference pressure, and 5000 ps of steps. At last, the production MD of the protein and protein–ligand complexes was run for 10 ns with a time step interval of 2 fs. After successful completion of MDS, the MD trajectories were then used to calculate root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg) by using g_rms, g_rmsf, g_gyrate tools of GROMACS 5.0.7.
Binding free energy calculation using MM-PBSA
The free energy calculation provides a quantitative estimation of interactions between protein and ligand that help to understand the stability of that protein–ligand complex [18]. The binding free energy including the free solvation energy (polar and nonpolar solvation energies) and potential energy (electrostatic and Vander Waals interactions) of each protein–ligand complexes was calculated by the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) method. The MD trajectories were processed before doing MM-PBSA calculations for last 1 ns. The MM-PBSA binding free energy calculation was done with ‘g_mmpbsa’ [18] script. The binding energy is calculated by using the following equation:
where ΔG binding = the total binding energy of the complex, G receptor = the binding energy of free receptor, G ligand = the binding energy of unbounded ligand.
Result and discussion
The molecular docking, virtual screening (VS), the assessment of physicochemical properties, and bioavailability of lead compounds play a crucial role in searching of novel and potential lead molecules to the protein targets associated with human diseases [19, 20]. However, the feasibility of large-scale virtual screening mainly depends on deciding the accurate target, selection of suitable chemical compound datasets, and the critical assessment of pharmacokinetic profiles of lead molecules [21]. We employed virtual screening of compounds from lichen compounds library for the viral target Mpro involve in COVID-19. Molecular docking, drug-likeness and toxicity prediction, X-score, MDS, and MM-PBSA analysis of lead molecules showed two promising hits that can be evaluated as antiviral molecules to control the global health crisis of COVID-19 (Fig. 1).
Virtual screening
Before conducting the virtual screening, molecular docking protocol was validated by docking the reference ligand X77 into a binding pocket obtained from the crystal structure of target protein Mpro. The docked ligand was superimposed to compare with an experimental ligand. Usually, RMSD value is used to validate the docking protocol. The RMSD value between the experimental and docked X77 was 0.84 Angstrom, which is perfectly acceptable. The result revealed that the docked reference molecules, X77 (orange) was completely superimposed with that of co-crystallized X77 (blue) (Fig. 2b) and both experimental as well as docked X77 showed interaction with the same amino acid residues by hydrogen and hydrophobic bonds as found in the crystal structure of Mpro (Fig. 2c). Our docking protocol produced a similar docking pose of X77 which was in the crystal structure of Mpro. Thus, this protocol was considered good enough for reproducing the docking results similar to X-ray crystal structure and therefore can be applied for further docking experiments.
The virtual screening of all lichen compounds (n = 412) was performed by molecular docking in the active site of a target protein using AutoDock Vina. From molecular docking, a total of 27 compounds were screened which showed binding energy ranging from -13.8 to -8.3 kcal mol−1 against Mpro (Table 1). The binding energy of the reference molecule, X77 was − 8.2 kcal mol−1. All screened hits showed lower and significantly better binding energy against the target protein in comparison to the reference molecule. The molecular docking result suggests that screened compounds may have the same mechanism of action as the reference molecule. Then, all these 27 compounds and X77 were further used for drug-likeness prediction.
Drug-likeness analysis
It has been reported that drug molecules showing good binding affinity with the target protein may fail in a clinical trial at advanced stages of drug discovery due to lack of drug-likeness property [22]. Hence we analyzed the drug-likeness of screened compounds using DruLiTo software. DruLiTo can calculate more than 23 physicochemical properties which are important for evaluating the drug-likeness of a molecule. Here, the drug-likeness was measured under the empirical thumb rule of drug-likeness i.e. Lipinski rule of 5. According to Lipinski’s RO5, that most “drug-like” molecules have Log P ≤ 5, molecular weight ≤500, number of hydrogen bond acceptors ≤10, and number of hydrogen bond donors ≤5. Among the 27 compounds, six compounds, viz. calycin, acetylportentol, russulfoen, thelephoric acid, roccellin, and rhizocarpic acid, showed better pharmacokinetics and successfully passed in RO5 evaluation. Pan assay interference compounds (PAINS) are chemical compounds that likely to interfere in screening technologies via several means but particularly through protein reactivity because they tend to react nonspecifically with numerous biological targets rather than specifically affecting one desired target. PAINS remover was used to remove the PAINS from screened hits and for their exclusion in bioassays [12]. Out of six compounds that follow RO5, two compounds, Thelephoric acid and Roccellin were filtered out and the remaining four compounds successfully passed the PAINS filter. The compounds which show better pharmacokinetics and satisfy the fundamental RO5 and PAINS filter are accepted as drug-like molecules. As per the RO5 and PAINS filter, the drug-likeness result of hit compounds is shown in Table 2.
Toxicity risk prediction
The US Food and drug administration toxicity risk predictor tool OSIRIS server used to predict the toxicity of screened compounds [23]. OSIRIS predicted various toxicity risk properties such as tumorigenicity, mutagenicity, irritation, and reproductive development toxicity. The results of toxicity prediction for all four hit compounds are summarized in Table 3. The drug-score show ranges between 0 and 1, where the value 1 indicates the good possibility of a compound to be drug molecule, whereas, the score value 0 indicates that compounds having no possibilities of drug candidates. The toxicity test shows that the reference molecule, X77 and two compounds, Calycin and Rhizocarpic acid have no risk of toxicity and remaining two compounds were toxic. Acetylportentol showed a high risk of Irritant effect while Russulfoen showed a high risk of tumorigenicity, mutagenicity, irritation, and reproductive toxicity.
Rescoring of docking results
X-Score is normally used to validate the binding energies of the protein–ligand complex obtained from docking. Thus, we used X-Score to re-score the binding energies of screened hits. The X-Score associated binding energy of the reference compound and screened hits against the target protein is compiled in Table 4. The X-Score results validated the molecular docking results as Rhizocarpic acid shows a better X-Score with Mpro than Calycin. Both the screened hits show the comparable value of X-Score as the reference molecule, X77.
Thus, considering the molecular docking, drug-likeness, and toxicity prediction results, we found Calycin and Rhizocarpic acid may be exploited as promising drug candidates for the development Glnt of antiviral drug molecules against COVID-19.
Visualization
PyMOL was used to visualize the 3D interactions of the protein–ligand complex. The docked poses of screened two compounds with Mpro is shown in Fig. 3a, b. Calycin forms one hydrogen bonds having distance 2.8 Å with Glu166 (Fig. 3a), while another compound Rhizocarpic acid form five hydrogen bonds with Mpro, 3 H-bond with Glu166 having distance 2.0 Å, 2.4 Å, and 2.8 Å, and one H-bond each with Gln189 and Glb192 with 2.3 Å and 2.5 Å bond distance respectively (Fig. 3b). The reference molecule, X77 also found to interact with Glu166 of Mpro through hydrogen bond (Fig. 2c). According to protein–ligand interaction, Calycin and Rhizocarpic acid bind with the active site residues Threonine and Glutamine (Glu166 and Gln189) of Mpro protein, and therefore, these two hit compounds may inhibit the Mpro of SARS-CoV-2.

Docked poses of the top hit compounds (Orange stick), a calycin and b rhizocarpic acid with Mpro. Mpro is in blue color cartoon representation. Active site residues are in black colored lines. Hydrogen bonds that are formed in between protein and compound are shown by Green lines and bond length as red dotted lines. 2D Interactions of X77 and screened compounds Calycin (c) and Rhizocarpic acid (d) with the active site of Mpro. The ligand structure is represented as thick purple stick in the center and the binding site residues involved in the hydrophobic interaction are depicted with the red half-circle, hydrogen bond showed by the green dotted line and the red circle shows the active site residues
Further, to get insights into the binding mechanism of the screened compounds in the active sites of the Mpro, we performed 2D interactions analysis of the docked complexes by LigPlot + v.1.4.5 software as shown in Fig. 3c and d. The reference molecule, X77, show interaction with several residues via significant interactions, including hydrogen and hydrophobic interactions. It forms three hydrogen bonds with Gly143, His163, and Glu166 of 3.24 Å, 3.09 Å, and 2.75 Å, respectively, and 12 hydrophobic bonds with Thr25, His41, Phe140, Leu141, Asn142, Ser144, Cys145, His164, Met165, Asp187, Arg188, Gln189 and yields the binding energy − 8.3 kcal mol−1 by AutoDock Vina and − 9.67 by X-Score (Fig. 2c). Calycin formed a hydrogen bond with active site residues Glu166 of 3.16Å. In addition, Mpro-Calycin complex make hydrophobic interaction with His41, Met49, Phe140, Leu141, Asn142, Ser144, Cys145, His163, Met165, Asp187, Arg188, Gln189 and gives the binding energy − 8.4 kcal mol−1 by AutoDock Vina and 8.47 by X-Score (Fig. 3c). Rhizocarpic acid shows a low-energy complex with Mpro as indicated by Autodock Vina and X-Score, i.e., − 8.7 kcal·mol−1 and − 9.11 kcal·mol−1, respectively. It also formed interaction with active site residues. It formed three hydrogen bonds with Glu166, Gln189, Gln192 of 3.32 Å, 2.98 Å, 3.27 Å and also yielded hydrophobic interactions with His41, Met49, Phe140, Leu141, Asn142, Ser144, Cys145, His163, His164, Met165, Pro168, Asp187, Arg188, Thr190 of Mpro (Fig. 3d).
From the analysis of molecular interaction between protein–ligand complexes, we observed that most of the hit compounds show common interaction and are involved in H-bond interaction and hydrophobic interaction with the same residue as shown in Table 5 which suggested the crucial role of hydrogen and hydrophobic interactions to hold the ligand at the active site of the target protein.
Molecular dynamic simulation (MDS)
The MDS was performed for predicting the stability of the screened seven hits. Three systems (Mpro-X77 (reference), Mpro-Calycin, and Mpro-Rhizocarpic acid) were subjected to 10 ns MDS. The structural changes and dynamic behavior in complexes were analyzed by the various computational analyses like RMSD, RMSF, RG calculation, and values are shown in Table 6.
Root-mean-square deviation RMSD
To determine the conformational and structural stability of Mpro and Mpro-ligand complexes, the differences between the backbone atoms of native protein from initial conformation to its final position was monitored through RMSD analysis. The deviations that occurred during the simulation describe the stability of conformation. Smaller deviations in protein reflect its more stable nature. RMSD score for the C-alpha backbone was calculated for 10 ns simulation. Figure 4a shows the plot of RMSD (nm) vs. time (ns) for native protein Mpro, reference complex Mpro-X77, Mpro-Calycin, and Mpro-Rhizocarpic acid complexes. From this figure, we can see that all the complexes are stable and produced stable trajectories for further analysis. The average value of RMSD for protein and all the complexes is shown in Table 6. The average RMSD values for protein were 0.14 ± 0.02 nm while for complexes; Mpro-X77, Mpro-Calycin, and Mpro-Rhizocarpic acid were found to be 0.13 nm, 0.15 nm, and 0.17 nm, respectively, with standard deviation 0.02. Both the studied complexes showed a similar RMSD value as reference complex Mpro-X77 which confirmed the stability of both the complexes.

MD simulation studies. a RMSD, b RMSF, and c Radius of gyration as a function of time. In all systems, the color code indicate- Mpro protein (black), Mpro-X77 (red), Mpro-Calycin (green), and Mpro-Rhizocarpic acid (blue)
Root-mean-square fluctuation (RMSF)
The RMSF was calculated for predicting the motions of the Mpro and Mpro-ligand complexes. The RMSF value can describe changes in the conformation of the protein due to binding with ligand during MDS. The rigid structures of protein like helix and sheets show low RMSF value, while loose structures containing region like sheets and turns show higher RMSF value. The RMSF plot of Mpro and all Mpro-ligand complexes is shown in Fig. 4b. RMSF plot shows that the secondary conformations of Mpro remain stable during the simulation of 10 ns. The average RMSF values for Mpro protein, Mpro-X77, Mpro-Calycin, and Mpro-Rhizocarpic acid complexes were recorded as 0.07, 0.08, 0.08, and 0.09 nm, respectively, (Table 6). Both the complexes showed similar average RsMSF value as compared to the Mpro and Mpro-X77 reference complex and represented these are very stable complex and does not cause much fluctuation after binding. The RMSF results represented that both predicted complexes were stable, and hence, these predicted compounds have the potential to inhibit the catalytic activity of Mpro.
Radius of gyration (RG)
The radius of gyration (Rg) shows the level of compactness in the structure of the protein due to presence or absence of ligands. The time evolution plot of Rg for Mpro protein and all Mpro-ligand complexes is shown in Fig. 4c. The average Rg value for Mpro protein, Mpro-X77, Mpro-Calycin, and Mpro-Rhizocarpic acid were found to be 1.84 nm, 1.88 nm, 1.75 nm, and 1.84 nm respectively (Table 6). The Mpro-Calycin complex showed much less Rg value as compared to the Mpro protein and other complexes, suggesting that it forms a more compact and stable complex as compared to other systems, though another hit showed relatively good Rg value similar to reference complex. From Table 6, it is visible that both the systems exhibited relatively similar and consistent Rg values of the reference which indicates that these are perfectively superimposed with each other, and they exhibit similar compactness and stability as reference. This showed that all complexes achieved relatively stable folded conformation during the 10 ns trajectory of MD simulation at the constant temperature of 300 K and the constant pressure of 1 atm. Overall, it can be concluded that the complexation of protein with hit compounds increases the compactness/rigidity of the Mpro structure, leading to increased overall stability.
Binding free energy calculation using MM-PBSA
The free energy analysis using MM-PBSA is applied to validate the docking energy of the protein–ligand complex. It was carried out using a python script MmPbSaStat.py provided in the g_mmpbsa package. The calculations of binding free energies were performed using the 1 ns of MD trajectories. The results of MM-PBSA are represented in Table 7. The Mpro-X77, Mpro-Calycin, and Mpro-Rhizocarpic acid complexes showed, − 91.78 kJ mol/1, − 42.42 kJ mol/1, and − 57.85 kJ mol/1 binding free energies, respectively. The binding free energy analysis showed that both Mpro-ligand complex(s) were stable. It confirms that both selected small molecules (Calycin and Rhizocarpic acid) can bind efficiently at the binding site of Mpro protein and could be used as lead compounds.
To identify the key residues involved in ligand binding toward protein, per residue interaction energy profile was created using the MM‐PBSA approach (Fig. 5) for the last 1 ns of MD trajectories. For a clear depiction of the results, only the active site residues are shown in Fig. 5. From the plot, it was revealed that Thr25, Leu27, Met49, Phe140, Leu141, Ser144, Cys145, His163, Met165, and Asp187 were the actively participating amino acid residues in both the predicted hits. The per residue interaction profile showed that most of the residues showed a negative binding affinity, while few residues showed a positive binding affinity. The residues that showed a negative binding affinity played an important role in stabilizing the protein‐ligand complex. Active site residues Thr25, Met49, Cys145, and Met165 showed higher binding affinity as compared to other residues. The results revealed that Thr25, Met49, Cys145, and Met165 play an important role in protein–ligand stabilization.

The contributions of individual amino acid residues of Mpro to the total binding energies of Mpro-ligand complexes. In all systems, the color code indicates- Mpro-X77 (Black), Mpro-Calycin (Red), and Mpro-Rhizocarpic acid (Green). Negative values indicate a stabilization effect on Mpro-ligand interactions
Conclusion
COVID-19 becomes a global concern, due to widespread outbreaks and lack of treatment. Therefore, it is necessary to find and evaluate treatment methods more quickly. In this case, computational methods are very effective and helpful. In this study, we employed various computational methods like virtual screening, drug-likeness analysis, toxicity prediction, MDS and MM-PBSA analysis for the identification of novel hit molecules as potential inhibitors for Mpro, the protein belongs to COVID-19. Here, we used a broad library of lichen compounds for screening purposes. Based on molecular docking, and binding affinity a total 27 hit molecules were selected as lead compounds against Mpro. Using the extensive pharmacokinetic drug-likeness analysis we obtained four compounds which followed Lipinski’s RO5 and PAINS filter. The X-Score, and toxicity analysis predicted two non-toxic compounds as they did not show any mutagenicity, tumorigenicity, and other effects and gave better binding affinity toward Mpro. Finally from the MDS and binding free energy results, we concluded that Calycin and Rhizocarpic acid are the best stable compounds that showed excellent binding affinities with Mpro. These observations suggested that these lichen compounds may be explored as a novel lead molecule for the rapid development of suitable drug candidates against COVID-19.
References
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506
Ingolfsdottir K (2002) Usnic acid. Phytochemistry 61(7):729–736
Molnar K, Farkas E (2009) Current results on biological activities of lichen secondary metabolites: a review. Z Naturforsch, C: J Biosci 65(3–4):157–173
Esimone CO, Grunwald T, Nworu CS, Kuate S, Proksch P, Uberla K (2009) Broad spectrum antiviral fractions from the lichen Ramalina farinacea (L.). Ach Chemother 55(2):119–126
Fazio AT, Adler MT, Bertoni MD, Sepulveda CS, Damonte EB, Maier MS (2007) Lichen secondary metabolites from the cultured lichen mycobionts of Teloschistes chrysophthalmus and Ramalina celastri and their antiviral activities. Z Naturforsch, C: J Biosci 62(7–8):543–549
Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A (2020) Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 Billion compounds. Mol Inform
De Clercq E (2002) Strategies in the design of antiviral drugs. Nat Rev Drug Discov 1(1):13–25
Huneck S, Yoshimura I (1928) Identification of lichen substances. Springer, Berlin, pp 29–46
Joshi T, Sharma P, Joshi T, Chandra S (2019) In silico screening of anti-inflammatory compounds from Lichen by targeting cyclooxygenase-2. J Biomol Struct Dyn, 1–19
Gohlke H, Hendlich M, Klebe G (2000) Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol 295(2):337–356
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44(1):235–249
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53(7):2719–2740
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47(7):1750–1759
Wang R, Lai L, Wang S (2002) Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des 16(1):11–26
Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854
Joshi T, Joshi T, Sharma P, Chandra S, Pande V (2020) Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. J Biomol Struct Dyn, pp 1–18
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD Jr (2009) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962
Mishra CB, Kumari S, Prakash A, Yadav R, Tiwari AK, Pandey P, Tiwari M (2018) Discovery of novel methylsulfonyl phenyl derivatives as potent human cyclooxygenase-2 inhibitors with effective anticonvulsant action: design, synthesis, in silico, in vitro and in vivo evaluation. Eur J Med Chem 151:520–532
Peterson B, Weyers M, Steenekamp JH, Steyn JD, Gouws C, Hamman JH (2019) Drug bioavailability enhancing agents of natural origin (Bioenhancers) that modulate drug membrane permeation and pre-systemic metabolism. Pharmaceutics 11(1)
Verma P, Tiwari M, Tiwari V (2018) In silico high-throughput virtual screening and molecular dynamics simulation study to identify inhibitor for AdeABC efflux pump of Acinetobacter baumannii. J Biomol Struct Dyn 36(5):1182–1194
Hoelder S, Clarke PA, Workman P (2012) Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol 6(2):155–176
Mabkhot YN, Alatibi F, El-Sayed NN, Al-Showiman S, Kheder NA, Wadood A, Rauf A, Bawazeer S, Hadda TB (2016) Antimicrobial activity of some novel armed thiophene derivatives and petra/osiris/molinspiration (POM) analyses. Molecules 21(2)
Acknowledgement
The authors acknowledge the Department of Botany, Kumaun University, S.S.J Campus, Almora and Rashtriya Uchchatar Shiksha Abhiyan (RUSA) program for providing basic facilities to conduct this research work.
Funding
There was no funding source to carry out this research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no competing interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Joshi, T., Sharma, P., Joshi, T. et al. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol Divers 25, 1665–1677 (2021). https://doi.org/10.1007/s11030-020-10118-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-020-10118-x