
Abstract
There is substantial clinical and experimental evidence that ammonia is a major factor in the pathogenesis of hepatic encephalopathy. In the article is demonstrated that in hepatocellular dysfunction, ammonia detoxification to glutamine (GLN) in skeletal muscle, brain, and likely the lungs, is activated. In addition to ammonia detoxification, enhanced GLN production may exert beneficial effects on the immune system and gut barrier function. However, enhanced GLN synthesis may exert adverse effects in the brain (swelling of astrocytes or altered neurotransmission) and stimulate catabolism of branched-chain amino acids (BCAA; valine, leucine, and isoleucine) in skeletal muscle. Furthermore, the majority of GLN produced is released to the blood and catabolized in enterocytes and the kidneys to ammonia, which due to liver injury escapes detoxification to urea and appears in peripheral blood. As only one molecule of ammonia is detoxified in GLN synthesis whereas two molecules may appear in GLN breakdown, these events can be seen as a vicious cycle in which enhanced ammonia concentration activates synthesis of GLN leading to its subsequent catabolism and increase in ammonia levels in the blood. These alterations may explain why therapies targeted to intestinal bacteria have only a limited effect on ammonia levels in patients with liver failure and indicate the needs of new therapeutic strategies focused on GLN metabolism. It is demonstrated that each of the various treatment options targeting only one the of the ammonia-lowering mechanisms that affect GLN metabolism, such as enhancing GLN synthesis (BCAA), suppressing ammonia production from GLN breakdown (glutaminase inhibitors and alpha-ketoglutarate), and promoting GLN elimination (phenylbutyrate) exerts substantial adverse effects that can be avoided if their combination is tailored to the specific needs of each patient.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatic encephalopathy (HE) results from impaired detoxification capacity of the liver leading to accumulation of various substances that may have deleterious effects on the function of the central nervous system. A key role in the development of HE has generally been ascribed to ammonia. Accumulation of ammonia in the brain results in alterations in activities of important enzymes, swelling of astrocytes, redistribution of cerebral blood flow and metabolism from cortical to sub-cortical areas, and imbalance of neural transmission (Bosoi and Rose 2009; Felipo and Butterworth 2002).
Although it is clear that the major cause of HE is the accumulation of ammonia, and various treatment strategies are at the clinician’s disposal, many patients with liver injury die due to the resulting hepatic coma. Current treatment of HE is mainly aimed at reducing the production and intestinal absorption of ammonia by antibiotics, and non-absorbable disaccharides. However, there is plenty of evidence that the main source of ammonia is not intestinal bacteria and that the crucial role in pathogenesis of hyperammonemia has glutamine (GLN) catabolism in enterocytes that is delivered to the intestine by the blood stream (Olde Damink et al. 2002; Nance and Kline 1971).
The first aim of this article is to review alterations in GLN synthesis and breakdown in liver failure and their relationship to the development of hyperammonemia and HE. The second aim is to discuss positive and negative aspects of various treatment strategies of hyperammonemia focused on GLN metabolism.
GLN and ammonia metabolism in the physiological state
GLN is unique amino acid serving as a “nitrogen shuttle” among organs, as a preferred respiratory fuel for rapidly proliferating cells, such as immune cells and enterocytes, a regulator of acid base balance through production of urinary ammonia, and playing important signaling role in many process, including expression of genes, cytokine and hormone production, and cell proliferation. During stress illness the body’s requirements for GLN frequently exceed the individual’s ability to synthesize sufficient amounts of this amino acid (Hardy and Hardy 2008; Wernerman 2011). Therefore, GLN is classified as a “conditionally essential” amino acid in the critically ill, the replacement of which is a rational therapeutic approach in GLN-deficient states.
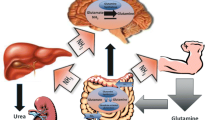
There is ingenious cooperation of various tissues in the synthesis and breakdown of GLN in order to keep low (<40 μmol/l) concentrations of ammonia in the systemic circulation and to deliver carbon and nitrogen to rapidly proliferating cells (Fig. 1). GLN is synthesized de novo from glutamate and ammonia in a process catalyzed by glutamine synthetase (GS):

The role of GLN in ammonia detoxification in physiological state. The most of endogenous GLN is synthesized from glutamate and ammonia in the brain, skeletal muscle, and likely the lungs. GLN released to the circulation is catabolized in enterocytes, kidneys, and liver to ammonia, and this is detoxified to urea in the liver or excreted together with urea in the urine
GS is present in almost all tissues. Substantial activity is in the brain, heart, and perivenous hepatocytes. However, the main GLN producer in the body is skeletal muscle because of its large mass relative to other GS containing organs (Huizenga et al. 1996). Appreciable GS activity and the potential to release GLN have also the lungs; however, their contribution to whole body GLN production in humans is not clear (Cooper and Freed 2005).
GLN released from the skeletal muscle and brain into circulation is metabolized predominantly in enterocytes, immunocytes, periportal hepatocytes, and in the kidneys in the glutaminase (GA) reaction to ammonia and glutamate. The latter may be metabolized to α-ketoglutarate (α-KG) and ammonia by the glutamate dehydrogenase (GDH) reaction. Thus, two ammonia molecules may be produced from one molecule of GLN:
Ammonia produced in the intestine is carried by portal blood to the liver, where most of it is extracted by the periportal hepatocytes and detoxified in the urea cycle. Ammonia not utilized in ureagenesis is detoxified by perivenous hepatocytes through amidation of glutamate in the GS reaction. The importance of hepatic extraction of gut-derived ammonia is demonstrated by the fact that ammonia concentrations in systemic circulation are 5–20 times lower than that of portal blood (Walker 2009).
The kidneys take up GLN from the blood at an amount that equals 10–15 % of whole-body GLN flux by both the luminal and antiluminal tubule surfaces and subsequently distributes the ammonia produced from GLN between the urine and renal vein (van de Poll et al. 2004). Physiologically, approximately 70 % of the ammonia generated in the kidneys is released to the renal vein, and the remainder is excreted in urine (van de Poll et al. 2004). Presumably, most of the ammonia that appears physiologically in the systemic circulation is derived from GLN metabolism in the kidneys.
GLN and ammonia metabolism in liver injury
When the capacity of the liver to detoxify ammonia in the urea cycle is not sufficient, GLN synthesis from ammonia in extrahepatic tissues is activated. Elevated concentrations of GLN can be detected in the blood and in various tissues, especially in disorders of the urea cycle (Maestri et al. 1992) and in acute liver failure (Clemmesen et al. 2000). The stimulatory effect of hyperammonemia on GLN synthesis has been demonstrated in skeletal muscle (Clemmesen et al. 2000; Girard and Butterworth 1992; Holecek et al. 2011) and in the brain (Norenberg et al. 2005). The lungs may also be important, because their convenient anatomical position may remove gut-derived ammonia from the blood before its entrance to the systemic circulation.
The studies dealing with the effects of GLN supplementation have demonstrated that enhanced GLN availability increases both GA activity in enterocytes and the portal load of ammonia (Salloum et al. 1990; Souba et al. 1990). Patients with liver cirrhosis exert increased intestinal GA activity that correlates with minimal HE (Romero-Gómez et al. 2004). Enhanced GLN availability also activates ammonia production in the kidneys (Tietze et al. 1992; Gougoux et al. 1985) and it is clear that the kidneys serves to lower the blood ammonia concentration during hyperammonemia by increasing urine ammonia excretion (Owen et al. 1961). Unfortunately, the capability of the kidneys to enhance the urinary excretion of ammonia is limited. During severe liver insufficiency, ammonia disposal capacity appears to be exceeded and the kidneys increase ammonia release to the renal veins (Dejong et al. 1993).
These findings of enhanced ammonia detoxification in tissues with high GS activities and enhanced GLN breakdown to ammonia in tissues with high GA activity suggests activation of GLN-ammonia cycling among tissues and that the main source of ammonia in patients with liver disease is catabolism of GLN of endogenous origin in enterocytes and the kidneys (Fig. 2).

The role of GLN in ammonia detoxification in liver failure. In liver failure, ammonia escapes the urea cycle and is detoxified to GLN in the brain, skeletal muscle, and lungs. Enhanced GLN availability leads to enhanced GLN catabolism to ammonia in enterocytes and the kidneys. Thus GLN-ammonia cycling among tissues is activated. PSS Portal-systemic shunts
As only one molecule of ammonia is detoxified to GLN in GLN synthetase reaction, whereas two ammonia molecules may appear from gradual breakdown of GLN in GA and GD reactions in the gut and the kidneys, these events can be seen as a vicious cycle. Enhanced ammonia concentration due to impaired detoxification to urea in the liver activates synthesis of GLN in skeletal muscle and in the brain leading to enhanced GLN catabolism to ammonia in enterocytes and the kidneys, and to subsequent increase in ammonia levels in the blood (Fig. 3).

Supposed vicious cycle in GLN synthesis and breakdown in pathogenesis of hyperammonemia in liver injury. Enhanced ammonia concentration due to impaired detoxification to urea in the liver activates GLN synthesis in skeletal muscle and in the brain leading to enhanced GLN catabolism to ammonia in enterocytes and the kidneys, and to subsequent increase in ammonia levels in the blood
Adverse effects of enhanced ammonia detoxification to GLN
Enhanced rates of GLN synthesis in muscle, brain, and probably in the lungs is undoubtedly a useful response that enables the body, at least temporarily, to suppress the rise of ammonia in liver failure. Beside ammonia detoxification, enhanced GLN production may provide other beneficial effects. There is evidence that GLN is an essential fuel for enterocytes and that enhanced GLN availability may be of benefit for the barrier function of the intestine (Meritt et al. 1989). Enhanced GLN levels may also favorably affect the immune system, glucose production in the kidneys, and protein balance in skeletal muscle (Stumvoll et al. 1999). Unfortunately, enhanced rates of ammonia detoxification to GLN may exert serious adverse effects–especially in the brain and skeletal muscle.
Adverse effects in the brain
The role of alterations of GLN metabolism induced by hyperammonemia in pathogenesis of HE has been reviewed in a number of excellent articles (Norenberg et al. 2005; Albrecht et al. 2010; Lockwood et al. 1984; Suárez et al. 2002). In short, in the brain, there is a neuron-glial metabolic interaction known as glutamate-GLN cycle that maintains low extracellular concentrations of glutamate and protects neurons from the deleterious effects of ammonia (Daikhin and Yudkoff 2000). Hyperammonemia enhances ammonia extraction by the brain, increases GS activity in astroglial cells located in glutamatergic areas, and thus leads to enhanced accumulation of GLN in astrocytes (Fig. 4). There is strong evidence that GLN accumulation in astrocytes contributes to the cerebral edema observed in acute hyperammonemia and to the presence of Alzheimer type II astrocytes in chronic hyperammonemia (Albrecht et al. 2010; Dolman et al. 1988). In addition, enhanced GLN synthesis may alter the availability of glutamate, an excitatory amino acid neurotransmitter and, due to stimulatory effect of GLN on the uptake of large neutral amino acids across the blood–brain barrier (Cangiano et al. 1983), participate in the pathogenesis of brain amino acid imbalance reported in patients with HE (Fischer and Baldessarini 1971).

Effects of hyperammonemia on GLN metabolism in the brain. Enhanced GLN synthesis may cause edema in astrocytes, alterations in availability of excitatory neurotransmitter glutamate, and amino acid imbalance in the brain. AA Amino acids; GA Glutaminase; GLN Glutamine; Glu Glutamate; GS Glutamine synthetase
Adverse effects in skeletal muscle
Hyperammonemia significantly increases GS activity and GLN synthesis from glutamate in skeletal muscle (Girard and Butterworth 1992). To ensure the continuous detoxification of ammonia to GLN, it is inevitable that glutamate becomes more available. The main source of amino nitrogen for synthesis of glutamate from α-ketoglutarate (α-KG) in skeletal muscle is branched-chain amino acids (BCAA; valine, leucine, and isoleucine). Because the BCAA aminotransferase reaction is reversible and near equilibrium, enhanced glutamate consumption activates glutamate synthesis from α-KG and BCAA catabolism (Fig. 5). The loss of BCAA from cytosol is compensated by their influx from extracellular fluid, presumably by the exchange of BCAA with GLN via L-transporter system (Meier et al. 2002).

Effects of hyperammonemia on GLN and BCAA metabolism in skeletal muscle (Holecek et al. 2011). Hyperammonemia enhances GLN synthesis that may enhance the consumption of BCAA, resulting in their deficiency in extracellular fluid. BCAA deficiency may participate in the pathogenesis of muscle wasting and HE. 1 branched-chain aminotransferase; 2 branched-chain α-ketoacid dehydrogenase; 3 GLN synthetase. ECF The extracellular fluid; BCAA Branched-chain amino acids; BCKA Branched-chain keto acids; BCA-CoA Branched-chain acyl-CoA; αnKG α-ketoglutarate; Glu Glutamate; Ala Alanine
These metabolic alterations explain the pathogenesis of the BCAA deficiency in liver cirrhosis that was enigmatic for many years. Enhanced rates of BCAA catabolism have been demonstrated in ammonia infused and cirrhotic rats, and in patients with liver cirrhosis (Leweling et al. 1996; Holecek et al. 1996b; Holecek et al. 2000; McCullough et al. 1992). Recently, clear evidence that hyperammonemia decreases the BCAA concentration in extracellular fluid was provided using isolated skeletal muscle (Holecek et al. 2011).
Together with increased levels of aromatic amino acids (phenylalanine, tyrosine, and tryptophan), decreased levels of BCAA in blood plasma have been implicated in the “false neurotransmitter” hypothesis as a possible cause of hepatic encephalopathy. Fischer and Baldessarini postulated that due to the decreased concentrations of BCAA, the aromatic amino acids flood the central nervous system and cause an imbalance in the synthesis of dopamine, noradrenaline and serotonin and/or may cause formation of “false neurotransmitters” such as octopamine, phenylethanolamine and tyramine (Fischer and Baldessarini 1971).
Therapeutic perspectives
The data mentioned above demonstrate the importance of GLN in the pathogenesis of hyperammonemia in liver injury and that some effects of enhanced GLN-ammonia cycling among tissues are apparently beneficial, but others are detrimental.
Positive effects: | - ammonia detoxification - improves gut barrier function - improves the function of immune system |
Negative effects: | - ammonia production from GLN in the gut and kidneys - swelling of astrocytes, altered neurotransmission - decrease of BCAA in extracellular fluid |
The existence of a vicious cycle in which GLN produced from ammonia in skeletal muscle and brain is used for synthesis of ammonia in enterocytes and the kidneys may explain why traditional gut-targeted therapies of HE to suppress ammonia production by bacteria in the colon, such as non-absorbable disaccharides and antimicrobial drugs, have only limited effects (Als-Nielsen et al. 2004). Therefore, theoretically, to develop effective strategies for treating of hyperammonemia and HE, the good effects of enhanced GLN-ammonia cycling should be supported and the evil effects eliminated. This could be achieved by various nutritional and pharmacological interventions.
GLN restriction
GLN administration to patients with liver cirrhosis raises the level of ammonia in the blood. This phenomenon is used as an oral GLN challenge test for the diagnosis of subtle neurological deficits and to predict the risk of episodes of HE in the condition referred to as minimal HE (Oppong et al. 1997; Ortiz et al. 2005). Therefore, GLN intake in patients with liver disease and HE should be restricted to suppress ammonia production in the gut. When the diet is otherwise adequate, the organism may synthesize large amounts of GLN unless the patient is severely malnourished and has a very low muscle mass. However, as GLN content is high in most food, its restriction is applicable only in patients fed by artificial diet formulas.
Glutamate
The first study reporting success in relieving coma using glutamic acid was published by Walshe in 1953, who demonstrated a return of consciousness after sodium glutamate administration in five episodes of coma in three patients with subacute or chronic liver injury (Walshe 1953). Since then, several reports have appeared on the use of glutamic acid, glutamate, and/or gamma-ethylester of glutamic acid in the treatment of high blood ammonia levels with conflicting results (McDermott et al. 1955; Santagati et al. 1979; Marchina et al. 1979).
It has been assumed that the effect of glutamate in the treatment of HE lies in its key role in binding ammonia to form GLN (Walshe 1953). However, there are studies demonstrating that the intestinal mucosa preferentially metabolizes glutamate as a source of energy and very small changes in plasma glutamate have occurred after adding large quantities of glutamate to the food (Stegink et al. 1982; Tsai and Huang 2000). Therefore, more likely is that oral glutamate administration decreases blood ammonia indirectly by sparing catabolism of endogenous GLN in enterocytes. This suggestion is supported by findings of decreased intestinal GA activity and increased plasma GLN concentrations after monosodium glutamate supplementation (Boutry et al. 2011).
α-Ketoglutarate
α-Ketoglutarate (α-KG) is the precursor of glutamate and GLN in the intermediate metabolism. When 14C(U) α-KG was parenterally administered to rats, radioactivity was recovered in GLN in the kidneys, intestinal mucosa, and skeletal muscle (Vaubourdolle et al. 1988). The rationale for α-KG in the treatment of hyperammonemia is also subnormal α-KG levels in patients with urea cycle disorders and portal-systemic encephalopathy (Batshaw et al. 1980). Of particular note is the extensive oxidation of α-KG by enterocytes that may suppress GLN catabolism and ammonia production by intestinal cells (Yao et al. 2012). The adverse side effect of α-KG administration may be catabolism of BCAA as demonstrated by increased levels of branched-chain keto acids in hemodialysis patients (Riedel et al. 1996).
Unfortunately, there are no studies evaluating the effect of α-KG in patients with hyperammonemia. Parenteral α-KG prevented the decrease in GLN in skeletal muscle after surgery (Hammarqvist et al. 1991). The decrease in ammonia levels (but without beneficial effects on the treatment of coma) was demonstrated after ornicetil (ornithine-α-KG) infusion in various forms of hepatic disease by Chainuvati et al. (1977).
BCAA
The peak of research in the field of BCAA use in HE treatment belongs to the period of extensive work of Fischer’s group, who demonstrated that BCAA administration normalizes amino acid imbalance in plasma, thus reducing the influx of aromatic amino acids (phenylalanine, tyrosine, and tryptophan) to the brain, and consequently improving alterations in neurotransmitter synthesis (Fischer and Baldessarini 1971; Fischer et al. 1976). Unfortunately, the enthusiasm for the BCAA dwindled in 1990s, when large clinical trials failed to confirm the efficacy of BCAA in HE treatment (Als-Nielsen et al. 2003). An updated systematic review on the effects of oral BCAA compared with control supplements or placebo for patients with cirrhosis and recurrent overt or minimal HE showed that BCAA supplements improve manifestations of HE but have no effect on survival (Gluud et al. 2013).
It can be hypothesized that BCAA might be more effective in patients with marked depression of BCAA. However, a serious problem in many trials evaluating the effectiveness of BCAA is the lack of information about alterations in aminoacidemia. An increase in BCAA levels has been observed in acute injury or in the case of acute decompensation of liver disease (Rosen et al. 1977; Hehir et al. 1985; Holecek et al. 1996a; Holecek et al. 1999).
The BCAA may also ameliorate HE by their positive effects on protein balance and stimulatory effects on ammonia detoxification to GLN in skeletal muscle (Ruderman and Berger 1974; Odessey et al. 1974; Jia et al. 2013). Unfortunately, enhanced ammonia production from GLN breakdown in the intestine and the kidneys may later exert harmful effects on the development of hepatic encephalopathy (Holecek 2013). The increase in blood ammonia after BCAA supplementation has been demonstrated both in patients with cirrhosis and healthy subjects (MacLean et al. 1996; Dam et al. 2011).
GA inhibitors
GA inhibitors are expected to preferentially block ammonia generation by GA in the intestine that seems to be the main cause of systemic hyperammonemia in cirrhotics (Romero-Gómez et al. 2004). A promising compound is THDP-17, which inhibited GA in cell cultures of colon carcinoma (Diaz-Herrero et al. 2012). However, GA inhibitors have potentially harmful effects on the function of enterocytes and immune cells that use GLN not only for synthesis of nucleic acids but also as important respiratory fuel (Windmueller and Spaeth 1980; Tjader et al. 2007). Therefore, GA inhibitors should reduce GA activity only partially. Favorable effects may obtain with simultaneous administration of GA inhibitors and α-KG that may replace GLN as an energy source in enterocytes (Yao et al. 2012).
Phenylbutyrate/phenylacetate
After ingestion, phenylbutyrate is rapidly oxidized to phenylacetate, which is conjugated to GLN to yield phenylacetylglutamine in the liver and kidneys. Phenylacetylglutamine is then quantitatively excreted by the urine. Removal of GLN from the body may suppress ammonia production from GLN and promote ammonia detoxification in the GS reaction. Sodium phenylbutyrate is effective in the clinical management of hyperglutaminemia and hyperammonemia in patients with urea cycle disorders (Brusilow 1991; Enns et al. 2007). A new investigational agent for treating of HE containing no sodium and having favorable palatability is glycerol phenylbutyrate. The results of clinical studies suggest that glycerol phenylbutyrate can be administered safely to cirrhotic subjects and has the potential to lower blood ammonia (McGuire et al. 2010).
The obvious risk of enhanced GLN removal from the body by phenylbutyrate/phenylacetate treatment is GLN depletion, which may exert adverse effects on the body, such as impaired immune response and gut integrity. GLN depletion may also activate BCAA catabolism and suppress BCAA levels as observed in phenylbutyrate-treated patients with urea cycle disorders (Scaglia et al. 2004) and thus exert deleterious effects on the course of HE development.
As a novel treatment of hyperammonemia and HE that seems to have a more complex way of action is combined administration of ornithine and phenylacetate (OP). Ornithine is believed to increase ammonia fixation into GLN and GLN is subsequently thought to react with phenylacetate forming phenylacetylglutamine which is excreted in urine (Jalan et al. 2007). In bile duct ligated cirrhotic rats, OP treatment reduced arterial ammonia concentration and increased that of GLN 30 min after treatment but not after 15 h (Dadsetan et al. 2013). A progressive drop in ammonia and GLN in blood plasma and a rise of phenylacetlyglutamine in urine were demonstrated when OP was administered to cirrhotic patients after an episode of upper gastrointestinal bleeding (Ventura-Cots et al. 2013).
Conclusions
Hepatocellular dysfunction results in impaired hepatic clearance of ammonia that is detoxified to GLN in the skeletal muscle, brain, and likely the lungs. Enhanced GLN production may exert beneficial effects on immune system and gut barrier function in addition to its ability to detoxify ammonia. Conversely, adverse effects may be observed in the brain (swelling of astrocytes and altered neurotransmission) and in skeletal muscle (catabolism of BCAA).
The majority of GLN released from the skeletal muscle and the brain to the circulation is catabolized in enterocytes and the kidneys back to the ammonia, which, in the case of liver injury, escapes detoxification to urea, appears in peripheral blood, and exerts stimulatory effects on GLN synthesis in the skeletal muscle and brain as noted above. Enhanced GLN-ammonia cycling among tissues can be seen as a vicious cycle in which enhanced ammonia detoxification to GLN in skeletal muscle and in the brain leads to enhanced GLN catabolism to ammonia in enterocytes and the kidneys, and to subsequent rise in ammonia levels in the blood (Fig. 3).
The alterations in GLN metabolism induced by liver injury are sensitive to various nutritional and pharmacological interventions that may be exploited in the prevention and treatment of HE. Unfortunately, targeting only one ammonia-lowering mechanism, such as enhancing GLN synthesis (BCAA), suppressing ammonia production (GA inhibitors and α-ketoglutarate), and/or promoting GLN elimination (phenylbutyrate) may exert substantial adverse effects (Table 1).
Theoretically, the problem of adverse side effects of individual ammonia-lowering strategies can be avoided by using a combination tailored to the specific needs of each patient.
Rational combinations in cirrhotic patients with high ammonia and low BCAA levels might be BCAA (correction of BCAA deficiency and stimulation of ammonia detoxification to GLN), α-KG (suppression of GLN breakdown to ammonia in the gut and substitution of α-KG consumed in synthesis of glutamate), and phenylbutyrate (increase in GLN removal). In patients with acute liver failure, combine GA inhibitors and phenylbutyrate with α-KG (α-KG may replace GLN as an energy substrate and thus avoid adverse effects of GA inhibitors on the function of intestinal and immune cells). BCAA levels in patients with acute injury are normal or even increased, and their enhanced intake should be avoided.
Favorable effects of ammonia-lowering strategies have been reported in urea cycle disorders (Brusilow 1991; Enns et al. 2007) and in liver cirrhosis (Walshe 1953; McGuire et al. 2010). However, the use of ammonia-lowering strategies has not been found to be effective enough in patients with acute liver failure (Acharya et al. 2009; Bémeur and Butterworth 2013). In acute liver failure, there is evidence of a robust neuroinflammatory response characterized by microglial activation and increased brain production of pro-inflammatory cytokines that act synergistically with ammonia in pathogenesis of HE (Bémeur and Butterworth 2013). Systemic inflammatory response and signs of neuroinflammation occur also in chronic liver failure (Butterworth 2011). Therefore, careful studies are needed to evaluate the effects of combining of ammonia-lowering strategies with therapies reducing neuroinflammation such as antibiotics, N-acetylcysteine, indomethacin, etanercept, and minocycline.
In conclusion, recent discoveries about the role of activated GLN-ammonia cycling among tissues in the pathogenesis of HE are opening new treatment options focused on GLN metabolism that might be combined with the classical methods of inhibiting ammonia production by bacteria in the colon. An important task for clinical researchers seems to be performing well-conducted trials that would suggest treatment strategies that are most likely to succeed. Beside the effects on ammonia levels and HE development, special attention should be given to their potential adverse effects on GLN and BCAA homeostasis that may negate their favorable effects.
Abbreviations
- α-KG:
-
Alpha-ketoglutarate
- Ala:
-
Alanine
- BCAA:
-
Branched-chain amino acids
- GA:
-
Glutaminase
- GDH:
-
Glutamate dehydrogenase
- GLN:
-
Glutamine
- Glu:
-
Glutamate
- GS:
-
Glutamine synthetase
- HE:
-
Hepatic encephalopathy
- OP:
-
Ornithine and phenylacetate
References
Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK (2009) Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 136:2159–2168
Albrecht J, Zielińska M, Norenberg MD (2010) Glutamine as a mediator of ammonia neurotoxicity: a critical appraisal. Biochem Pharmacol 80:1303–1308
Als-Nielsen B, Koretz RL, Kjaergard LL, Gluud C (2003) Branched-chain amino acids for hepatic encephalopathy. Cochrane Database Syst Rev 2:CD001939
Als-Nielsen B, Gluud LL, Gluud C (2004) Non-absorbable disaccharides for hepatic encephalopathy: systematic review of randomised trials. BMJ 328(7447):1046
Batshaw ML, Walser M, Brusilow SW (1980) Plasma alpha-ketoglutarate in urea cycle enzymopathies and its role as a harbinger of hyperammonemic coma. Pediatr Res 14:1316–1319
Bémeur C, Butterworth RF (2013) Liver-brain proinflammatory signalling in acute liver failure: role in the pathogenesis of hepatic encephalopathy and brain edema. Metab Brain Dis 28:145–150
Bosoi CR, Rose CF (2009) Identifying the direct effects of ammonia on the brain. Metab Brain Dis 24:95–102
Boutry C, Bos C, Matsumoto H, Even P, Azzout-Marniche D, Tome D, Blachier F (2011) Effects of monosodium glutamate supplementation on glutamine metabolism in adult rats. Front Biosci 3:279–290
Brusilow SW (1991) Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res 29:147–150
Butterworth RF (2011) Neuroinflammation in acute liver failure: mechanisms and novel therapeutic targets. Neurochem Int 59:830–836
Cangiano C, Cardelli-Cangiano P, James JH, Rossi-Fanelli F, Patrizi MA, Brackett KA, Strom R, Fischer JE (1983) Brain microvessels take up large neutral amino acids in exchange for glutamine. Cooperative role of Na + −dependent and Na + −independent systems. J Biol Chem 258:8949–8954
Chainuvati T, Plengvanit U, Viranuvatti V (1977) Ornicetil on encephalopathy. Effect of ornicetil (ornithine alpha-ketoglutarate) on encephalopathy in patients with acute and chronic liver disease. Acta Hepatogastroenterol (Stuttg) 24:434–439
Clemmesen JO, Kondrup J, Ott P (2000) Splanchnic and leg exchange of amino acids and ammonia in acute liver failure. Gastroenterology 118:1131–1139
Cooper AJ, Freed BR (2005) Metabolism of [13N]ammonia in rat lung. Neurochem Int 47:103–108
Dadsetan S, Sørensen M, Bak LK, Vilstrup H, Ott P, Schousboe A, Jalan R, Keiding S, Waagepetersen HS (2013) Interorgan metabolism of ornithine phenylacetate (OP)—a novel strategy for treatment of hyperammonemia. Biochem Pharmacol 85:115–123
Daikhin Y, Yudkoff M (2000) Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr 130:1026S–1031S
Dam G, Keiding S, Munk OL, Ott P, Buhl M, Vilstrup H, Bak LK, Waagepetersen HS, Schousboe A, Møller N, Sørensen M (2011) Branched-chain amino acids increase arterial blood ammonia in spite of enhanced intrinsic muscle ammonia metabolism in patients with cirrhosis and healthy subjects. Am J Physiol Gastrointest Liver Physiol 301:G269–G277
Dejong CH, Deutz NE, Soeters PB (1993) Renal ammonia and glutamine metabolism during liver insufficiency-induced hyperammonemia in the rat. J Clin Invest 92:2834–2840
Díaz Herrero MM, del Campo JA, Carbonero P, Jover M, Vega Perez J, Iglesias Guerra F, Periñán I, Bautista JD, Romero Gómez M (2012) THDP-17 inhibits the glutaminase activity in Caco-2 cell cultures. THDP-17 inhibits the glutaminase activity in Caco-2 cell cultures. Gastroenterol Hepatol 6(S11):7, abstract
Dolman CL, Clasen RA, Dorovini-Zis K (1988) Severe cerebral damage in ornithine transcarbamylase deficiency. Clin Neuropathol 7:10–15
Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A (2007) Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med 356:2282–2292
Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67:259–279
Fischer JE, Baldessarini RJ (1971) False neurotransmitters and hepatic failure. Lancet ii:75–80
Fischer JE, Rosen HM, Ebeid AM, James JH, Keane JM, Soeters PB (1976) The effect of normalization of plasma amino acids on hepatic encephalopathy in man. Surgery 80:77–91
Girard G, Butterworth RF (1992) Effect of portacaval anastomosis on glutamine synthetase activities in liver, brain, and skeletal muscle. Dig Dis Sci 37:1121–1126
Gluud LL, Dam G, Borre M, Les I, Cordoba J, Marchesini G, Aagaard NK, Risum N, Vilstrup H (2013) Oral branched-chain amino acids have a beneficial effect on manifestations of hepatic encephalopathy in a systematic review with meta-analyses of randomized controlled trials. J Nutr 143: 1263–1268
Gougoux A, Vinay P, Halperin ML (1985) Regulation of renal ammoniagenesis in the dog with chronic metabolic acidosis: effect of a glutamine load. Am J Physiol 249:F745–F752
Hammarqvist F, Wernerman J, von der Decken A, Vinnars E (1991) Alpha-ketoglutarate preserves protein synthesis and free glutamine in skeletal muscle after surgery. Surgery 109:28–36
Hardy G, Hardy IJ (2008) Can glutamine enable the critically ill to cope better with infection? JPEN J Parenter Enteral Nutr 32:489–491
Hehir DJ, Jenkins RL, Bistrian BR, Wagner D, Moldawer LL, Young VR (1985) Abnormal phenylalanine hydroxylation and tyrosine oxidation in a patient with acute fulminant liver disease with correction by liver transplantation. Gastroenterology 89:659–663
Holecek M (2013) Branched-chain amino acids and ammonia metabolism in liver disease: Therapeutic implications. Nutrition. doi:10.1016/j.nut.2013.01.022
Holecek M, Mraz J, Tilser I (1996a) Plasma amino acids in four models of experimental liver injury in rats. Amino Acids 10:229–241
Holecek M, Tilser I, Skopec F, Sprongl L (1996b) Leucine metabolism in rats with cirrhosis. J Hepatol 24:209–216
Holecek M, Skalska H, Mraz J (1999) Plasma amino acid levels after carbon tetrachloride induced acute liver damage. A dose–response and time-response study in rats. Amino Acids 16:1–11
Holecek M, Sprongl L, Tichy M (2000) Effect of hyperammonemia on leucine and protein metabolism in rats. Metabolism 49:1330–1334
Holecek M, Kandar R, Sispera L, Kovarik M (2011) Acute hyperammonemia activates branched-chain amino acid catabolism and decreases their extracellular concentrations: different sensitivity of red and white muscle. Amino Acids 40:575–584
Huizenga JR, Gips CH, Tangerman A (1996) The contribution of various organs to ammonia formation: a review of factors determining the arterial ammonia concentration. Ann Clin Biochem 33:23–30
Jalan R, Wright G, Davies NA, Hodges SJ (2007) L-Ornithine phenylacetate (OP): a novel treatment for hyperammonemia and hepatic encephalopathy. Med Hypotheses 69:1064–1069
Jia H, Takahashi S, Saito K, Kato H (2013) DNA microarray analysis identified molecular pathways mediating the effects of supplementation of branched-chain amino acids on CCl(4) -induced cirrhosis in rats. Mol Nutr Food Res 57:291–306
Leweling H, Breitkreutz R, Behne F, Staedt U, Striebel JP, Holm E (1996) Hyperammonemia-induced depletion of glutamate and branched-chain amino acids in muscle and plasma. J Hepatol 25:756–762
Lockwood AH, Bolomey L, Napoleon F (1984) Blood–brain barrier to ammonia in humans. J Cereb Blood Flow Metab 4:516–522
MacLean DA, Graham TE, Saltin B (1996) Stimulation of muscle ammonia production during exercise following branched-chain amino acid supplementation in humans. J Physiol 493:909–922
Maestri NE, McGowan KD, Brusilow SW (1992) Plasma glutamine concentration: a guide in the management of urea cycle disorders. J Pediatr 121:259–261
Marchina MM, Renzi G, Serofilli S (1979) Medical treatment of hyperammoniemia in the elderly. Controlled clinical study. Minerva Med 70:811–818
McCullough AJ, Mullen KD, Kalhan SC (1992) Body cell mass and leucine metabolism in cirrhosis. Gastroenterology 102:1325–1333
McDermott WV, Wareham J, Riddell AG (1955) Treatment of hepatic coma with L-glutamic acid. N Engl J Med 253:1093–1102
McGuire BM, Zupanets IA, Lowe ME, Xiao X, Syplyviy VA, Monteleone J, Gargosky S, Dickinson K, Martinez A, Mokhtarani M, Scharschmidt BF (2010) Pharmacology and safety of glycerol phenylbutyrate in healthy adults and adults with cirrhosis. Hepatology 51:2077–2085
Meier C, Ristic Z, Klauser S, Verrey F (2002) Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J 21:580–589
Meritt J, Witkowski TA, Nagele R, Norcross ED, Stein TP (1989) Glutamine and smooth muscle morphology of the gut in rats on total parenteral nutrition. J Am Coll Nutr 8:537–544
Nance FC, Kline DG (1971) Eck’s fistula encephalopathy in germfree dogs. Ann Surg 174:856–862
Norenberg MD, Rao KV, Jayakumar AR (2005) Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis 20:303–318
Odessey R, Khairallah EA, Goldberg AL (1974) Origin and possible significance of alanine production by skeletal muscle. J Biol Chem 249:7623–7629
Olde Damink SW, Jalan R, Redhead DN, Hayes PC, Deutz NE, Soeters PB (2002) Interorgan ammonia and amino acid metabolism in metabolically stable patients with cirrhosis and a TIPSS. Hepatology 36:1163–1171
Oppong KN, Al-Mardini H, Thick M, Record CO (1997) Oral glutamine challenge in cirrhotics pre- and post-liver transplantation: a psychometric and analyzed EEG study. Hepatology 26:870–876
Ortiz M, Jacas C, Córdoba J (2005) Minimal hepatic encephalopathy: diagnosis, clinical significance and recommendations. J Hepatol 42:S45–S53
Owen EE, Johnson JH, Tyor MP (1961) The effect of induced hyperammonemia on renal ammonia metabolism. J Clin Invest 40:215–221
Riedel E, Nündel M, Hampl H (1996) alpha-Ketoglutarate application in hemodialysis patients improves amino acid metabolism. Nephron 74:261–265
Romero-Gómez M, Ramos-Guerrero R, Grande L, de Terán LC, Corpas R, Camacho I, Bautista JD (2004) Intestinal glutaminase activity is increased in liver cirrhosis and correlates with minimal hepatic encephalopathy. J Hepatol 41:49–54
Rosen HM, Yoshimura N, Hodgman JM, Fischer JE (1977) Plasma amino acid patterns in hepatic encephalopathy of differing etiology. Gastroenterology 72:483–487
Ruderman NB, Berger M (1974) The formation of glutamine and alanine in skeletal muscle. J Biol Chem 249:5500–5506
Salloum RM, Souba WW, Fernandez A, Stevens BR (1990) Dietary modulation of small intestinal glutamine transport in intestinal brush border membrane vesicles of rats. J Surg Res 48:635–638
Santagati G, Zibetti G, Forgione F, Spaghi A (1979) Anti-ammonemic activity of the gamma-ethyl ester of glutamic acid. Controlled clinical study. Minerva Med 70:595–603
Scaglia F, Carter S, O’Brien WE, Lee B (2004) Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab 81:S79–S85
Souba WW, Herskowitz K, Salloum RM, Chen MK, Austgen TR (1990) Gut glutamine metabolism. JPEN J Parenter Enteral Nutr 14:45S–50S
Stegink LD, Filer LJ Jr, Baker GL (1982) Plasma and erythrocyte amino acid levels in normal adult subjects fed a high protein meal with and without added monosodium glutamate. J Nutr 112:1953–1960
Stumvoll M, Perriello G, Meyer C, Gerich J (1999) Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int 55:778–792
Suárez I, Bodega G, Fernández B (2002) Glutamine synthetase in brain: effect of ammonia. Neurochem Int 41:123–142
Tietze IN, Sørensen SS, Eiskjaer H, Thomsen K, Pedersen EB (1992) Tubular handling of amino acids after intravenous infusion of amino acids in healthy humans. Nephrol Dial Transplant 7:493–500
Tjader I, Berg A, Wernerman J (2007) Exogenous glutamine–compensating a shortage? Crit Care Med 35:S553–S556
Tsai PJ, Huang PC (2000) Circadian variations in plasma and erythrocyte glutamate concentrations in adult men consuming a diet with and without added monosodium glutamate. J Nutr 130:1002S–1004S
van de Poll MC, Soeters PB, Deutz NE, Fearon KC, Dejong CH (2004) Renal metabolism of amino acids: its role in interorgan amino acid exchange. Am J Clin Nutr 79:185–197
Vaubourdolle M, Jardel A, Coudray-Lucas C, Ekindjian OG, Agneray J, Cynober L (1988) Metabolism and kinetics of parenterally administered ornithine and α-ketoglutarate in healthy and burned animals. Clin Nutr 7:105–111
Ventura-Cots M, Arranz JA, Simón-Talero M, Torrens M, Blanco A, Riudor E, Fuentes I, Suñé P, Soriano G, Córdoba J (2013) Safety of ornithine phenylacetate in cirrhotic decompensated patients: an open-label, dose-escalating, single-cohort study. J Clin Gastroenterol. doi:10.1097/MCG.0b013e318299c789
Walker V (2009) Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes Metab 11:823–835
Walshe JM (1953) The effect of glutamic acid on the coma of hepatic failure. Lancet 1(6770):1075–1077
Wernerman J (2011) Glutamine supplementation. Ann Intensive Care 1:25
Windmueller HG, Spaeth AE (1980) Respiratory fuels and nitrogen metabolism in vivo in small intestine of fed rats. Quantitative importance of glutamine, glutamate, and aspartate. J Biol Chem 255:107–112
Yao K, Yin Y, Li X, Xi P, Wang J, Lei J, Hou Y, Wu G (2012) Alpha-ketoglutarate inhibits glutamine degradation and enhances protein synthesis in intestinal porcine epithelial cells. Amino Acids 42:2491–2500
Acknowledgments
Supported by the program PRVOUK P37/02.
Conflict of interest
The author declares that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Holecek, M. Evidence of a vicious cycle in glutamine synthesis and breakdown in pathogenesis of hepatic encephalopathy–therapeutic perspectives. Metab Brain Dis 29, 9–17 (2014). https://doi.org/10.1007/s11011-013-9428-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-013-9428-9