
Abstract
Acinetobacter baumannii is notorious for causing serious infections of the skin, lungs, soft tissues, bloodstream, and urinary tract. Despite the overwhelming information available so far, there has still been no approved vaccine in the market to prevent these infections. Therefore, this study focuses on developing a rational vaccine design using the technique of epitope mapping to curb the infections caused by A. baumannii. An outer membrane protein with immunogenic potential as well as all the properties of a good vaccine candidate was selected and used to calculate epitopes for selection on the basis of a low percentile rank, high binding scores, good immunological properties, and non-allergenicity. Thus, a 240 amino-acid vaccine sequence was obtained by manually joining all the epitopes in sequence-wise manner with the appropriate linkers, namely AAY, GPGPG, and EAAAK. Additionally, a 50S ribosomal protein L7/L12, agonist to the human innate immune receptors was attached to the N-terminus to increase the overall immune response towards the vaccine. As a result, enhanced overall protein stability, expression, immunostimulatory capabilities, and solubility of the designed construct were observed. Molecular dynamic simulations revealed the compactness and stability of the polypeptide construct. Moreover, molecular docking exhibited strong binding of the designed vaccine with TLR-4 and TLR-9. In-silico immune simulations indicated an immense increment in T-cell and B-cell populations. Bioinformatic tools also significantly assisted with optimizing codons which allowed for successful cloning of constructs into desired host vectors. Using in-silico tools to design a vaccine against A. baumannii demonstrated that this construct could pave the way for successfully combating infections caused by multidrug-resistant bacteria.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Acinetobacter baumannii belongs to the Moraxellaceae family of gammaproteobacteria. With a genome size of between 30 and 40 kb, it is characterized as a Gram-negative, non-fermenting, and rod-shaped organism (Howard et al. 2012). A. baumannii is considered rather a new pathogen as it caused its first successful outbreak during the Iraq war in the early 20th century and thus, is also referred to as “Iraqibacter” (Scott et al. 2007). Consisting of a highly plastic genome, it is considered to be the most dangerous microbe of the genus Acinetobacter, responsible for causing 80% of infections in healthcare settings (Manchanda et al. 2010). The bacteria has a remarkable capability to colonize and infect the host’s skin, tissues, blood, pulmonary tracts, central nervous system (CNS), and surgical wounds thus causes serious health implications which involve urinary tract infections, soft tissue infections, bloodstream infections (BSI), pulmonary infections, and meningitis (Karageorgopoulos and Falagas 2008; Howard et al. 2012). In the wake of increasing resistance to antibiotics, this pathogen has made it onto the WHO’s list of “the most dangerous microbes” (Xie et al. 2021). Although A. baumannii infections are not airborne, they can freely spread via contaminated equipment in hospitals or direct contact with infected persons or health care staff (Dijkshoorn et al. 2007). The risk of getting infected by A. baumannii is higher for immunocompromised elderly individuals, infants, patients who underwent antibiotic treatments recently, or patients who have been in hospitals for prolonged periods (Blanco et al. 2018). These infections could range from transient bacteremia to fulminant septic shock (Ballouz et al. 2017). The bloodstream infections are common among children: the most vulnerable victims of A. baumannii (Gramatniece et al. 2019). The overall mortality ratio of A. baumannii ranges from 23 to 68% (Morris et al. 2019).
In the light of increasing resistance rate of A. baumannii, the bacteria is able to develop resistance to almost all available antibiotics, leaving us with very few options of treatment (Benkő et al. 2020). In response, scientists are increasingly focusing on developing new preventive measures, out of which in-silico vaccine development has become a very popular methodology for managing dangerous pathogens. There has been a great technological advancement in the vaccine designing methods due to improvements in immunoinformatic and bioinformatics tools (Oli et al. 2020). A variety of approaches to identify novel vaccine candidates, such as structural vaccinology and reverse-vaccinology (RV) have immensely boosted the rate of vaccine-designing (Yang et al. 2021). Targeting suitable vaccine candidates from the genome of bacteria to instigate active and passive immune responses is a rational approach in mitigating the adversities caused by human pathogens. So far, a large number of antigens have been proposed as suitable vaccine candidates against A. baumannii which involve outer membrane porin A (OmpA) (Nie et al. 2020), NucAb (Garg et al. 2016), BamA (Singh et al. 2017), BauA (Sefid et al. 2015), Bap (Girija et al. 2021), TolB (Song et al. 2018), Ata (Bentancor et al. 2012), FilF (Singh et al. 2016), K1 polysaccharide (Russo et al. 2013), Phospholipase D (Zadeh Hosseingholi et al. 2014) and a few others. Despite the extensive knowledge of putative vaccine candidates, no A. baumannii vaccine has been approved by FDA or introduced into the market. The proposed antigens have many shortcomings including complex compositions, increased toxicity and safety concerns, solubility problems, poor cross-protectivity, and endotoxin contamination that hinder their translation into human therapeutics (Moriel et al. 2013; Ansari et al. 2019; Ren et al. 2019). To help overcome these problems, there is a need for the prediction of potent and safe epitopes to rationally construct multi-epitope subunit vaccine (MESV) that could effectively activate antibody as well as cell-mediated immune response. Epitopes could be predicted with high precision and accuracy using different bioinformatic servers (Soria-Guerra et al. 2015).
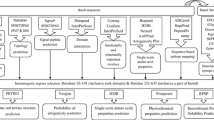
Among antigens, outer membrane porins (OMPs) have proved to be of significant importance as they are not only found in abundance but also readily detected by the immune system. In a recent study, an outer membrane protein (DcaP) has been suggested as an appropriate vaccine candidate in A. baumannii (Fereshteh et al. 2020). While the role of DcaP in conferring pathogenicity to A. baumannii needs to be explored, its crystal structure, localization, and expression have already been confirmed as a result of which it has been categorized as a “hijack target” for future antimicrobials (Bhamidimarri et al. 2019). As compared to other OMPs, little is known about DcaP protein and its immunogenic potential. This study is conducted to envisage immunogenic regions of this protein to design a MESV using different bioinformatic tools and approaches. A hierarchical approach to three-dimensional protein modeling was used to receive insights concerning the structure of the designed construct. Thereafter, rigorous evaluations of physicochemical aspects, protein stability, binding energies towards toll-like receptors (TLR-4 and TLR-9) and simulations for probable immune responses were performed. Finally, the MESV was ligated into Escherichia coli vector after codon optimization so that the overall protein expression could be assessed. A flowchart illustrating general steps followed throughout the study is shown in Fig. 1.

General steps followed throughout the study to design a multi-epitope vaccine (MEV) against Acinetobacter baumannii
Materials and Methods
Protein Sequence Retrieval
DcaP protein sequence of the reference strain ACICU of A. baumannii was retrieved from Genbank in the FASTA format (accession ID gb|ACC56846.1) (Benson et al. 2013). To determine the physical and chemical aspects of the selected protein, a web-based tool ExPASy ProtParam was employed (Artimo et al. 2012). The antigenicity of protein was calculated through the VaxiJen server v 2.0 (Doytchinova and Flower 2007). VaxiJen performs the antigenicity analysis of a protein using the information based on physicochemical properties. BlastP analysis was carried out to find conservation levels of the protein in A. baumannii as well as among different Acinetobacter strains.
Assessment of Cytotoxic T-Lymphocyte (CTL) Epitopes
NetCTL1.2 was utilized to locate 9-mer CTL epitopes in the selected vaccine candidate at a default threshold level of 0.75 (Larsen et al. 2007). Three factors determining the prediction efficiency of NetCTL 1.2 are (i) TAP transport efficiency (ii) peptides binding to MHC class 1 and (iii) proteasomal C-terminus cleavage. With the former, a weight matrix is used, while the latter two are achieved by artificial neural networks (ANN).
Assessment of Helper T-lymphocyte (HTL) Epitopes
For the purpose of obtaining information regarding the HTL epitopes related to a reference set of seven human HLA, an online webserver was utilized, IEDB MHC-II (Paul et al. 2013). The reference set of HLA included HLA-DRB3*01:01, HLA-DRB3*02:02, HLA-DRB1*07:01, HLA-DRB1*15:01, HLA-DRB1*03: 01, HLA-DRB4*01:01, and HLA-DRB5*01:01. HTLs are selected based on their binding capabilities towards alleles as depicted by IC50 scores in nanomoles. The values < 50 nM, < 500 nM and < 5000 nM refer to strong, moderate, and weak binding affinities of epitopes for MHC-II, respectively. Similarly, percentile rank, inversely related to the binding affinities, also aids in categorizing the epitopes by comparing them to large sets of sequences having the same length (Paul et al. 2013). An online tool IFN-epitope was employed to predict the HTL epitopes that specifically stimulate interferon-gamma cytokines (Kumar Pandey et al. 2018). The algorithm of this server uses a hybrid approach in which the strengths of motif and SVM models are combined.
Designing the Multi-epitope Subunit Vaccine (MESV) Construct
Carefully chosen high-affinity CTL and HTL epitopes were connected in a sequence-wise manner with the help of different connecting sequences known as “linkers” to construct the MESV. In the final construct, a total of four CTLs showing the highest combined score and three HTLs showing the lowest percentile rank were combined in such a way that the AAY linker separated the CTL epitopes whereas GPGPG linkers parted the HTL epitopes. AAY improves the epitope partition to make the C-terminus of CTLs readily available for molecule-mediated binding, which eventually increases the epitope presentation. Similarly, GPGPG linkers were used to fuse the HTLs to better stimulate the HTL immune response (Livingston et al. 2002). A commonly employed strategy to enhance the efficacy of the vaccine and to create a biased or a controlled immune response is to add an adjuvant (Liang et al. 2020). Therefore, a TLR4-inducing 50S ribosomal protein L7/L12 (Uniprot accession ID; P9WHE3) was used as an adjuvant and connected to the N-terminus of the vaccine construct using EAAK linker. The total length of the newly constructed vaccine construct was found out to be 240 amino acids.
Analysis of Allergenicity, Antigenicity Solubility, and Physicochemical Aspects
To make sure that the vaccine construct has the desired non-allergenic behavior, two online webservers AllergenFP v1.0 and AllerTop v2.0 were exploited (Dimitrov et al. 2014a, b). Both servers are the alignment-free method and determine the allergenic behavior of a protein by analyzing its amino acid properties. The antigenicity of the construct was determined with the help of the VaxiJen server (Doytchinova and Flower 2007). To avoid the cross-reactivity of protein, BLASTp analysis was carried out against the human proteome. Further analysis of the physicochemical properties was carried out via ExPASy web using which molecular weight, aliphatic index, instability index (II), grand average of the hydropathicity (GRAVY) value, theoretical pI, solubility, and estimated half-life of the designed construct were examined (Artimo et al. 2012). The solubility of the vaccine was cross-checked by using an online tool SOLpro available in the SCRATCH suite (Cheng et al. 2005). The secondary structure of the vaccine was determined using the PSIPRED v3.3 web server (McGuffin et al. 2000).
Generation of 3D Structure
3D structure of the designed construct was determined to better understand its biological function. For this purpose, I-TASSER (iterative Threading ASSEmbly Refinement) was used (Roy et al. 2010). I-TASSER generates 3D models of proteins on the basis of sequence to structure to function paradigm and has been ranked as one of the best servers for protein structure prediction.
Refinement, Validation, and Flexibility Analysis
After the initial refinement performed by ModRefiner (Xu and Zhang 2011), secondary refinements of the 3D structure were performed by using the GalaxyRefine server (Giardine et al. 2005). To validate the refinements being carried out, RAMPAGE and ProSA-web servers were employed to perform the Ramachandran plot analysis and Z-score analysis, respectively (Wiederstein and Sippl 2007). ERRAT webserver helped in locating possible potential anomalies that could be present in the structure (Colovos and Yeates 1993). To determine the flexibility of the designed MESV, CABS-Flex 2.0 server was utilized (Jamroz et al. 2013). It is a well-developed tool to effectively perform simulations of a protein’s structural flexibility.
Screening for B-Cell Epitopes (Linear and Conformational)
For a vaccine construct to incite a humoral response, it needs to have epitopes for B-cell lymphocytes in its regions (Tahir ul Qamar et al. 2020). To depict the linear as well as conformational B-cell epitopes, the Ellipro webserver was utilized (Ponomarenko et al. 2008). This tool makes use of algorithms to perform three tasks which include (i) determining PI (protrusion index) of residue, (ii) estimation of protein shape, and (iii) cluster formation of the neighboring residues.
Molecular Dynamic Simulation Analysis
Molecular dynamic simulations are necessary to stabilize the protein hence, GROMACS (GROningen MAchine for Chemical Simulations) software was employed for performing MD simulations. To track vaccine constructs’ behavior in realistic biological settings, GROMACS replicates the behaviors and environment of real-life cellular environments (Abraham et al. 2015). First, structural processing was done using the pdb2gmx command to generate a gro file compatible with the force field. Optimized Potential for Liquid Simulations-all atom (OPLS-AA) force field was applied to the structure and a rhombic box was defined in which the structure was placed in the center at 1.0 nm from the box edges and 2.0 nm from its periodic images (Kaminski et al. 2001). As a next step, the SPC/E water model having the force constant (Kpr) of 1000 kJ mol−1 nm−2 was used to fill the water into the defined system to create an aqueous environment. The total charge on the designed protein was determined and a tool for neutralizing the overall charge called “genion” was used. A total of ten solvent molecules were replaced by sodium ions. After the solvation and charge neutralization, energy minimization was carried out to ensure no steric clashes or abnormal geometry is encountered during the dynamics study. Xmgrace was utilized to view obtained results in the form of graphs. Equilibrations were performed carefully in two stages under isothermal–isochoric (or NVT) ensemble and isothermal–isobaric (or NPT) ensemble to stabilize the pressure, temperature, and density of the protein at 1000 ps. Post-equilibration trajectory analysis was done for 60 ns. Finally, RMSD of the backbone and RMSF of the protein were analyzed and demonstrated in the form of graphs.
Molecular Docking
The binding ability of the designed MESV was studied by carrying out the docking analysis with TLR-4 and TLR-9 which are well-known for inducing an immune response against Gram-negative bacteria especially A. baumannii (Kim et al. 2013), (Noto et al. 2015). ClusPro tool was employed to study protein-protein interactions (Kozakov et al. 2017). This web server performs docking using PIPER program which is based on a highly efficient Fast Fourier transform (FFT) correlation approach. PIPER symbolizes the interaction energy between two proteins using an equation.
where Erep and Eattr depict the energy contributions to the van der Waal’s forces in the form of repulsions or attractions. Eelec is a term for electrostatic energy and Edars represents free energy change due to the removal of water molecules from the system, whereas w1, w2, w3, and w4 are co-efficient which represent weight of the corresponding term (Vajda et al. 2017).The binding affinities between vaccine construct and the immune receptors were cross-verified using Patchdock webserver which uses a rigid body algorithm (Schneidman-Duhovny et al. 2005). The top 10 results generated by Patchdock were submitted to Firedock for further improvement (Andrusier et al. 2007).
Codon Optimization and Cloning
In order to express the gene of interest in a vector, it is necessary to have conformity between the codons of the host vector and the gene of interest so that high levels of activity are ensured at the time of purification. For this purpose, the Java Codon Adaptation Tool (JCat) was used, which corroborates codon compliance by optimizing the vaccine construct sequence (Grote et al. 2005). A protein must exhibit desirable GC content (30–70%) and CAI-values (> 0.8) to validate increased expression. The designed construct was optimized according to E. coli strain-K12, which is known for its quick reproduction rates and ability to survive (Biselli et al. 2020). Two restriction enzymes NdeI and XhoI were added at the N- and C-terminus of the protein to finally insert it into the pET28a (+) vector using the SnapGene 2.0.1 software.
Immune Response Simulation Analysis
C-ImmSim web server uses a position-specific scoring matrix (PSSM) which helps in understanding the magnitude of immune response generated as a result of vaccine dosage at different time intervals (Rapin et al. 2010). The most recommended period between two injections of vaccines is 4 weeks (Nain et al. 2020). Therefore, three injections at time steps of 1, 84, and 168 were administered, with all other settings set to default (Shey et al. 2019).
Results
Retrieval of Protein Sequence
The first step in the designed workflow was to obtain the protein sequence from the outer membrane of A. baumannii reference strain ACICU. The sequence was collected from Genbank in FASTA format (gb|ACC56846.1). This protein sequence was tested for its antigenic potential using the VaxiJen v 2.0 server. The antigenic value was found out to be 0.7 which is greater than the threshold value of 0.4. To further explore the physical and chemical aspects of protein, ExPASy Protparam webserver was employed (Supplementary Table S1).
Conservation of DcaP in Acinetobacter Genus
To assess the conservation levels, BlastP analysis of the outer membrane protein was performed, which showed high conservation levels based on 961 strains with 90 to 96% protein identity in A. baumannii strains and 86.8 to 94.6% in other species from the genus Acinetobacter (94.6% with A. nosocomialis, 91.32% with A. seifertii, 89.5% with A. calcoaceticus, 89.1% with A. lactucae, 88.52% with A. pitti, 88.51 with A. lactucae, 86.8% with A. oleivorans). A phylogenetic tree showing significant sequence identity is shown in Supplementary Fig. S1. This widespread conservation of DcaP demonstrates its broad-spectrum vaccine potential against Acinetobacter strains.
Cytotoxic T Cell Epitopes Mapping
NetCTL1.2 predicted 15 CTL epitopes (9-mer) for DcaP protein (Supplementary Table S2). Four epitopes GTFRVRHAY, NSNLTNTSY, GSISNGANY, and ATDAEVDAL which displayed antigenic, non-allergenic and non-toxic behavior were selected (Table 1).
Mapping of Helper T-Lymphocyte Epitopes
The IEDB MHC-II tool was used to evaluate 15-mer HTL epitopes against seven human allele reference sets (Supplementary Table S3). The epitope sequences with the lowest percentile rank that showed high binding affinities were finally selected for incorporation into the vaccine construct. Table 2 shows the selected epitopes and their properties.
Construction of the Multi-epitope Subunit Vaccine
VaxiJen analysis for the predicted epitopes was carried out to find the antigenicity of each peptide. A total of seven non-allergenic and highly antigenic epitopes from the outer membrane porin DcaP were selected, arranged, and coupled together with the help of linkers to create a linear vaccine construct. Four CTL epitopes were connected using the AAY linkers. HTL epitopes were joined with the help of GPGPG linkers. To enhance the immunogenic potential of the vaccine, the 50S ribosomal L7/L12 adjuvant (ID: P9WHE3) was linked to the N-terminus via the EAAAK linker. As a result, 240 amino acid long vaccine construct was designed (Fig. 2).

Illustration of the vaccine design. a Sequences of the selected CTL and HTL epitopes, with bracketed numerals showing their placement in the final vaccine construct. b Overview of the final vaccine construct with adjuvants, linkers, and epitopes
Inspection of Physical, Chemical, Behavioral, and Structural Aspects of the Vaccine
The proposed construct was confirmed to be non-allergen, non-toxic, non-reactogenic, and antigenic in behavior. BlastP analysis against human proteome suggested that there was no similarity of sequence to the humans. Also, the physicochemical analysis revealed that the MESV construct had a theoretical PI and a molecular weight of 4.7 and 25 kDa respectively. The GRAVY score was − 0.208, with a negative sign indicating the vaccine’s hydrophilic nature. The aliphatic index was 83.96 and the instability index was 20. This highlighted the protein’s ability to remain stable upon expression. The SOLpro website determined that protein was soluble after overexpression with a probability of 0.87. All these properties confirmed the usage of this construct as a potential vaccine candidate. PSIPRED v 3.3 estimated the secondary structure of the vaccine sequence. There were 17.5% beta strands, 42.5% alpha-helix, and 40% coil structure in the anticipated structure (Supplementary Fig S2).
Three-Dimensional Structure Prediction and Refinement
The top five 3D structures of vaccine, calculated by the I-TASSER webserver were predicted by using the top ten threading templates which showed alignment to the primary sequence (Fig. 3a). The normalized z-score of templates ranged from 0.26 to 5.62 representing good alignment with the query sequence. Confidence score (C-score) plays a fundamental role in determining the quality of a model with values differing from − 5 to 2. The more the value, the greater the credence in the structure. The predicted models had c-scores ranging from − 3.10 to − 4.62. The model with the highest c-score showing a TM score of 0.37 and RMSD of 13.1 Å was selected for further investigations (Fig. 3b). The TM score is used to determine how similar two protein structures are, and it eliminates variations caused by RMSDs. TM values more than 0.5 suggest correct topology, whereas scores less than 0.17 imply non-specific similarity (Zhang and Skolnick 2004).
The raw structure was sent to the ModRefiner first, and then to the GalaxyRefine website, to get a more refined structure. As a consequence, five models were created, with model 3 being chosen based on many characteristics including GDT-HA 0.9927, MolProbity 1.771, 97.1% Rama-favored, and RMSD value of 0.273. Ramachandran plot was analyzed using the RAMPAGE webserver. The improved model had a Ramachandran plot showing 98.32% of amino acids in the favorable region with no outliers (Fig. 3c).

MESV sequence, structure, and quality. a MESV sequence representation. b 3D structure of MESV colored-coded on the basis of secondary structure (red: beta strands, cyan: alpha helix, Magenta: coil). c Ramachandran plot exhibiting 98.32% in the Rama-favored region
Evaluation of 3D Structure and Flexibility
To ensure the good quality of structure, the refined structure was subjected to further validations using the ProSA-web and ERRAT webservers. ProSA-web predicted the Z-score of the protein to be − 6.57. ERRAT webserver was used to assess the potential errors in the 3D crude model that could compromise the global quality. Modeled protein’s quality factor was found out to be 95.53%. These scores indicated that our model has a high structural quality (Fig. 4).

MESV structural quality validation. a ProSA-web Z-score analysis showing the Z-score value of − 6.57. b ERRAT error prediction showing the quality of model as high as 95.53%
Furthermore, the flexibility of the vaccine was determined by an online tool CABS-flex 2.0, which performed 50 rounds of simulation at a default temperature of 1.4 °C. The collective model of 10 retrieved structures showed fewer fluctuations near the N-terminus as compared to the regions near the C-terminus (Fig. 5a). The generated contact map revealed contacts among different residues of all the ten final retrieved structures (Fig. 5b). Finally, the fluctuation plot represented the Root Mean Square Fluctuations (RMSF) of each amino acid from 0.0 to 5.3 Å (Fig. 5c). Such variations in the RMSF of the designed construct indicate its high flexibility and thus further reinforce its potential to be used as a probable vaccine.

Flexibility analysis of designed MESV. a Cartoon illustration of all ten models showing minimum fluctuations, predicted by CABS-flex. b Residue-residue interaction map. c RMSF-plot showing fluctuations ranging from 0.0 to 5.3 Å
Conformational and Linear B-Cell Epitopes Mapping
B-lymphocytes are critical for initiating pathogen-specific antibody-mediated responses (Hoffman et al. 2016). Therefore, an ideally designed vaccine construct should possess B-cell epitopes. Ellipro webserver was employed and default settings were used to predict linear and conformational B-lymphocyte epitopes. As a result, five linear and nine conformational stretches of BCEs were detected with antigenicity values ranging from 0.5 to 0.81 (Table 3) (Fig. 6).

Discontinuous B-cell Epitopes B-cell epitopes are shown in yellow color and protein construct in the form of grey sticks
Molecular Dynamic Simulation Analysis
MD simulation is a popular method for efficient analysis of structural reliability of proteins in a simulated environment which is closely related to realistic systems (Hansson et al. 2002). To further endorse the structural integrity of the vaccine construct, MD simulations were performed for 60 nanoseconds using a Linux-based molecular dynamics software, “GROMACS” (Abraham et al. 2015). In addition to the OPLS-AA force field, the SPC/E water model was used to solvate the protein. The net charge on the protein was found to be − 10, therefore 10 sodium ions were incorporated into the system by replacing the water molecules at positions 46378, 27898, 7537, 19234, 5188, 8170, 33283, 6562, 19885, and 41227 to neutralize the charge. As a result, only 14866 water molecules were left behind. To identify stable conformations, it is vital to minimize the global energy of molecules (Geng et al. 2019). Hence, energy minimization was performed for 500,000 steps. The steepest descents congregated after 567 steps, where the force attained was <1000 kJ mol−1. The GROMACS energies (temperature, pressure, and density) were analyzed using the Xmgrace and found to be desirable for further analysis (Table 4) (Fig. 7B–D). The resulting trajectories were analyzed to gain insights about the Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (Rg) of the construct. The stability and compactness of the structure during simulations are proven by the Rg graph (Fig. 7A). Calculated RMSD values of the backbone atoms were recorded to reach 0.36 nm and oscillate between 0.36 and 0.39 nm throughout the simulation, depicting the high stability of the protein (Fig. 7E). Furthermore, the computed RMSF values identified the regions with high flexibility, characterized by peak-like patterns (Fig. 7F).

MD simulation trajectory-based graphs for analysis of structural stability. Graphs generated by GROMACS at different stages of MD simulations of the designed construct. A Radius of gyration(RoG). The RoG of the vaccine construct stays compact, thereby supporting its stability during simulation. B Variations in temperature during simulation. During 1 ns, the temperature reached 300 K and fluctuated minimally. C Pressure plot. Average pressure of 0.362204 bar was achieved during 1 ns. D Graphical presentation of density during simulation. E The root mean square deviation of the backbone. RMSD plot shows that the RMSD of the peptide backbone is approximately 0.36 nm; meaning that it’s mostly maintained, indicating minimum structural deviations of the vaccine construct. F The root mean square fluctuation graph. RMSF plot illustrates the peak-like regions with a higher degree of flexibility
Molecular Docking of the Designed Construct with Toll-Like Receptors
Molecular docking of the designed MESV was carried out against TLR4 (PDB ID-4G8A) and TLR-9 (PDB ID-3WPF) using the ClusPro v 2.0 and Patchdock webservers. As a result, ClusPro v 2.0 generated 29 structures for each docking. The model that showed strong binding affinity and least intermolecular energy was selected in both cases. The lowest energy scores of − 1015 and − 1121 were observed during docking with TLR4 and TLR9 respectively (Figs. 8 and 9).

Molecular docking with TLR-4 and Interaction analysis. a Cartoon representation of the docked complex of TLR-4 (pale green) and designed vaccine (hot pink). b interacting amino acids depicted in black color. c schematic diagram of interactions between chains

Molecular docking with TLR-9 and Interaction analysis. a Cartoon representation of the docked complex of TLR-9 (marine blue) and designed vaccine (hot pink). b Interacting amino acids depicted in black color. c Schematic diagram of interactions between chains
The PatchDock webserver, following rigid-body docking, generated a list of 100 candidate complexes in the form of transformation files, which were then refined and scored again by FireDock. The output was a ranking of all the input solutions on the basis of global energy value. Based on the results of the designed construct, the highest global energy was − 46.27 and − 38.60 respectively for TLR-4 and TLR-9. Table 5 shows the global energy, energy contributed by hydrogen bond (HB) and Van der Waals forces (Attractive VdW) of the best docked complex.
Codon Optimization and Cloning
To carry out the expression analysis, codon optimization was followed by in-silico cloning of the designed vaccine construct inside the E. coli vector backbone. Codons in vaccine construct were tailored according to the codons in the E. coli expression system by using the JCat web tool. The generated cDNA sequence was found out to be 720 nucleotides in length (Supplementary Table S4). An ideal CAI value of 1 was obtained. Similarly, GC content (50.56%) was found to be desirable for achieving high protein expressions. 5` and 3` ends of the designed construct were restricted by using two restriction enzymes NdeI and XhoI, respectively. The restricted sequence was then successfully ligated into the pET28a (+) vector, which resulted in a cloned vector of 6043 bp. The cloned map was visualized and created using the SnapGene software (Fig. 10).

Cloned vaccine construct (red arrow) in PET28a (+) vector (black backbone) after codon optimization
Immune Simulations
By using C-ImmSim webserver, we assessed the vaccine’s ability to elicit an effective immune response if it were administered in a real-world setting. The results showed steady raises in the secondary and tertiary immune responses after the primary reaction. A considerable spike in levels of antibodies (IgM, IgM, IgM+ IgG, and IgG1+IgG2 antibodies) was observed (Fig. 11a). Similarly, a marked increase in B-cell numbers was also noted, in which IgG1 and IgM biotypes were detected along with significant memory cell formation (Fig. 11b). Upon receiving a secondary and tertiary injection dose, active T-cells were observed to significantly increase, but they progressively decreased at later stages (Fig. 11c, d). It is evident from Fig. 11e that upon vaccination, levels of dendritic cells (DCs) and macrophages increased prominently. Correspondingly, elevated amounts of cytokines (e.g., IL-2 and IFN-γ) were also noticed (Fig. 11f). These results indicate that the designed protein could generate positive immune responses.

In-silico immune simulations in the host in response to MESV administration. a Intermittent boosts in B-cell populations after secondary and tertiary doses. b Number of B-cell types in three successive immune responses. c Helper T-cell populations after three shots. d Cytotoxic T-cell populations after three vaccine administrations. e Dendritic cells per state. f Generation of cytokines and interleukins at three different stages
Discussion
A. baumannii infections occurring in lethal MDR forms have become a significant concern across the globe (Butler et al. 2019). Although numerous antimicrobial therapies are in pipeline for the treatment of these diseases, treatment of A. baumannii infections with vaccines has not been made achievable yet. Vaccines have proved to be the most efficient preventive tool against multiple infections and diseases (Rémy et al. 2015). Multi-epitope vaccines have an edge over classic vaccines due to their higher efficiency, fewer chances of cross-reactivity, cost-effectiveness, and ability to create biased immune responses (Sette et al. 2001; Goumari et al. 2019). Formerly, a plethora of multi-epitope vaccines has been conceived using computational tools against several bacterial infections such as Helicobacter pylori (Ghosh et al. 2021), Klebsiella pneumoniae (Dar et al. 2019), Pseudomonas aeruginosa (Solanki et al. 2019), Vibrio cholerae (Nezafat et al. 2016), Shigella spp. (Nosrati et al. 2019), Brucella (Saadi et al. 2017), Aeromonas hydrophilla (Bhattacharya et al. 2020), Prostate cancer (Patra et al. 2020) and many others. Considering the impact and efficacy of these vaccines, we followed a similar approach to design a multi-epitope based vaccine to thwart infections instigated by A. baumannii.
In the present study, we aimed to build a vaccine against A. baumannii based on antigenic epitopes extracted from the outer membrane protein DcaP of A. baumannii. DcaP is one of the most abundant precursors in the outer membrane and is involved in the catabolism of dicarboxylic acids (Bhamidimarri et al. 2019). This study differs from other similar studies on the basis of differently selected vaccine candidate having highly antigenic regions which show enhanced immunogenicity when tested in-silico. The Genbank sequence of the protein was retrieved and tested for its allergenic, toxic, and antigenic behavior. It was reported to be non-toxic, non-allergenic, and highly antigenic in nature. Therefore, it was considered further to map epitopes that could be incorporated into the vaccine.
As a next step, HTL and CTL epitopes were predicted using several webservers. HTLs play an important role in promoting humoral as well as cellular immunity (Alexander et al. 1998). Likewise, CTLs play a key role in developing the adaptive immunological response. CTL and HTL epitopes identification is the most important step in designing a suitable vaccine (Ashfaq et al. 2021). The selected epitopes were joined together with the help of proper linkers (AAY and GPGPG). Linkers have an important role in enhancing the epitope presentation, vaccine stability, and overall antigenicity (Dong et al. 2020).
To further help the construct to induce a strong immune response, a TLR-4 agonist adjuvant, 50S ribosomal subunit L7/L12 protein was connected to the N-terminus and attached to the epitopes with the help of EAAAK linker (Samad et al. 2020). Microbial products are actively recognized by human TLRs which as a result stimulate several immune responses (Ito et al. 2013). According to a study conducted by Kim et al., it has been proved that TLR-4 is far more active in generating an immune response as compared to other receptors (Kim et al. 2013). Similarly, TLR-9 has also shown active involvement against A. baumannii (Noto et al. 2015). Therefore, we carried out docking analysis of vaccine construct with TLR-4 and TLR-9. A thorough examination of the properties of the designed construct revealed that it was highly stable and capable of producing antibodies. It was found that 42.5%, 17.5%, and 40%, respectively, of the construct consisted of alpha helix, beta strand, and coil. In order to better comprehend the tertiary structure of MESV, the Ramachandran plot was analyzed, which reveals its probable conformation and ultimately helps to understand its stability as a whole (Gopalakrishnan et al. 2007). Generally, >98% residues in the Rama-favored region demonstrate the good quality of model. Our model exhibited 98.32% residues in the Rama-favored region. It gave a Z-score and ERRAT value of − 6.57 and 95.53%, respectively. The Z-score correlated with experimentally determined X-ray crystallographic structures, which gave credence to the good quality of the proposed structure. To ensure that the vaccine construct contains regions to which immunoglobulins could easily latch, we predicted the linear as well as conformational B-cell epitopes (Sanchez-Trincado et al. 2017). As a result, five linear and nine conformational stretches were found to be present in the construct which exhibited its capacity to generate enhanced humoral immune responses. The final sequence was subsequently codon adapted according to the codons used in the E. coli PET28a(+) vector for enhanced expression and purification. A CAI value of 1 and GC content of 50.56% were obtained from the optimized sequence indicating the exceptional levels of protein expression (Kar et al. 2020).
We evaluated the binding affinity and docking of TLR receptors (TLR4 and TLR9) with designed protein to see if the vaccine would bind to the receptors and form a stable MESV-TLR complex. The models showing the least intermolecular energies (− 1015 and − 1121) and highest binding affinities were selected and visualized with the help of PyMol software. The immune response generated by the human body was mimicked in-silico by using the C-ImmSim tool to understand the actual response. Generated results disclosed that immune responses were bolstered after secondary and tertiary vaccine injections. Many immunoglobulins belonging to different isotypes were also produced which helped in the development of memory cells. It could be seen from the results that the protein efficiently increases the populations of helper and cytotoxic T cells (Fig. 11c, d).
Vaccination using MESV has generated a lot of positive results in stimulating cellular as well as humoral immune responses against a plethora of microbial pathogens (Zhou et al. 2009; Nezafat et al. 2016; Hajighahramani et al. 2017; Dar et al. 2019; Solanki et al. 2019; Nain et al. 2020). Advancements in immunoinformatic approaches have led us to accurately determine immunogenic epitopes in a highly efficient, less time-consuming, and cost-effective manner (Tomar and De 2010; De Groot et al. 2020). To ensure the correctness of projected results, the data acquired in this study were analyzed using a variety of immunoinformatic techniques, algorithms, and databases. Hence, the designed vaccine construct could not only prompt cellular but also humoral immunity at increased levels and therefore may also be further subjected to high throughput in-vivo efficacy evaluations.
Conclusion
In conclusion, this study demonstrated the antigenicity of outer membrane porin DcaP from Acinetobacter baumannii by using its immune potentiating epitopes that simultaneously activate both arms of the human immune system. As a result of employing multiple immunoinformatic tools, the designed construct showed antigenic and non-allergenic properties, thereby reiterating the potential of the designed construct as immunogenic and putatively safe for therapeutic vaccine formulations. The predicted 3D structure of the construct further confirmed that it successfully binds to the human toll-like receptors (TLR-4 and TLR-9) and thus, efficiently triggers the cellular responses. Although we observed in-silico immune response of the designed chimeric vaccine in our study, the designed vaccine chimera needs further experimental validation to ensure its efficacy against A. baumannii infections.
Data Availability
All data includes within the manuscript.
References
Abraham MJ, Murtola T, Schulz R et al (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Alexander J, Fikes J, Hoffman S et al (1998) The optimization of helper T lymphocyte (HTL) function in vaccine development. Immunol Res 18:79–92. https://doi.org/10.1007/BF02788751
Andrusier N, Nussinov R, Wolfson HJ (2007) FireDock: fast interaction refinement in molecular docking. Proteins Struct Funct Genet 69:139–159. https://doi.org/10.1002/PROT.21495
Ansari H, Tahmasebi-Birgani M, Bijanzadeh M et al (2019) Study of the immunogenicity of outer membrane protein A (ompA) gene from Acinetobacter baumannii as DNA vaccine candidate in vivo. Iran J Basic Med Sci 22:669–675. https://doi.org/10.22038/ijbms.2019.30799.7427
Artimo P, Jonnalagedda M, Arnold K et al (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:W597–W603. https://doi.org/10.1093/nar/gks400
Ashfaq UA, Saleem S, Masoud MS et al (2021) Rational design of multi epitope-based subunit vaccine by exploring MERS-COV proteome: reverse vaccinology and molecular docking approach. PLoS ONE 16:e0245072. https://doi.org/10.1371/journal.pone.0245072
Ballouz T, Aridi J, Afif C et al (2017) Risk factors, clinical presentation, and outcome of Acinetobacter baumannii bacteremia. Front Cell Infect Microbiol 7:156. https://doi.org/10.3389/fcimb.2017.00156
Benkő R, Gajdács M, Matuz M et al (2020) Prevalence and antibiotic resistance of ESKAPE pathogens isolated in the emergency Department of a Tertiary Care Teaching Hospital in Hungary: a 5-Year retrospective survey. Antibiotics 9:624. https://doi.org/10.3390/antibiotics9090624
Benson DA, Cavanaugh M, Clark K et al (2013) GenBank. Nucleic Acids Res 41:D36–D42. https://doi.org/10.1093/NAR/GKS1195
Bentancor LV, Routray A, Bozkurt-Guzel C et al (2012) Evaluation of the trimeric autotransporter ata as a vaccine candidate against Acinetobacter baumannii infections. Infect Immun 80:3381–3388. https://doi.org/10.1128/IAI.06096-11
Bhamidimarri SP, Zahn M, Prajapati JD et al (2019) A multidisciplinary approach toward identification of antibiotic scaffolds for Acinetobacter baumannii. Structure 27:268–280.e6. https://doi.org/10.1016/j.str.2018.10.021
Bhattacharya M, Sharma AR, Sharma G et al (2020) Computer aided novel antigenic epitopes selection from the outer membrane protein sequences of Aeromonas hydrophila and its analyses. Infect Genet Evol 82:104320. https://doi.org/10.1016/J.MEEGID.2020.104320
Biselli E, Schink SJ, Gerland U (2020) Slower growth of Escherichia coli leads to longer survival in carbon starvation due to a decrease in the maintenance rate. Mol Syst Biol 16:e9478. https://doi.org/10.15252/msb.20209478
Blanco N, Harris AD, Rock C et al (2018) Risk factors and outcomes associated with multidrug-resistant Acinetobacter baumannii upon intensive care unit admission. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.01631-17
Butler DA, Biagi M, Tan X et al (2019) Multidrug resistant Acinetobacter baumannii: resistance by any other name would still be hard to treat. Curr Infect Dis Rep 21:1–17. https://doi.org/10.1007/s11908-019-0706-5
Cheng J, Randall AZ, Sweredoski MJ, Baldi P (2005) SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 33:W72–W76. https://doi.org/10.1093/nar/gki396
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2:1511–1519. https://doi.org/10.1002/pro.5560020916
Dar Z et al (2019) Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella Pneumoniae. Vaccines 7:88. https://doi.org/10.3390/vaccines7030088
De Groot AS, Moise L, Terry F et al (2020) Better epitope discovery, precision immune engineering, and accelerated vaccine design using Immunoinformatics tools. Front Immunol 11:442. https://doi.org/10.3389/fimmu.2020.00442
Dijkshoorn L, Nemec A, Seifert H (2007) An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. https://doi.org/10.1038/nrmicro1789
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP v.2—a server for in silico prediction of allergens. J Mol Model 20:1–6. https://doi.org/10.1007/s00894-014-2278-5
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014b) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30:846–851. https://doi.org/10.1093/bioinformatics/btt619
Dong R, Chu Z, Yu F, Zha Y (2020) Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front Immunol 11:1784. https://doi.org/10.3389/fimmu.2020.01784
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4. https://doi.org/10.1186/1471-2105-8-4
Fereshteh S, Abdoli S, Shahcheraghi F et al (2020) New putative vaccine candidates against Acinetobacter baumannii using the reverse vaccinology method. Microb Pathog 143:104114. https://doi.org/10.1016/j.micpath.2020.104114
Garg N, Singh R, Shukla G et al (2016) Immunoprotective potential of in silico predicted Acinetobacter baumannii outer membrane nuclease, NucAb. Int J Med Microbiol 306:1–9. https://doi.org/10.1016/j.ijmm.2015.10.005
Geng H, Chen F, Ye J, Jiang F (2019) Applications of molecular dynamics simulation in structure prediction of peptides and proteins. Comput Struct Biotechnol J 17:1162–1170. https://doi.org/10.1016/J.CSBJ.2019.07.010
Ghosh P, Bhakta S, Bhattacharya M et al (2021) A novel multi-epitopic peptide vaccine candidate against Helicobacter pylori: in-silico identification, design, cloning and validation through molecular dynamics. Int J Pept Res Ther 2021 272 27:1149–1166. https://doi.org/10.1007/S10989-020-10157-W
Giardine B, Riemer C, Hardison RC et al (2005) Galaxy: a platform for interactive large-scale genome analysis. Genom Res 15:1451–1455. https://doi.org/10.1101/gr.4086505
Girija ASS, Shoba G, Priyadharsini JV (2021) Accessing the T-cell and B-cell immuno-dominant peptides from A. baumannii biofilm associated protein (bap) as vaccine candidates: a computational approach. Int J Pept Res Ther 27:37–45. https://doi.org/10.1007/s10989-020-10064-0
Gopalakrishnan K, Sowmiya G, Sheik SS, Sekar K (2007) Ramachandran plot on the web (2.0). Protein Pept Lett 14:669–671. https://doi.org/10.2174/092986607781483912
Goumari MM, Farhani I, Nezafat N, Mahmoodi S (2019) Multi-epitope vaccines (MEVs), as a novel strategy against infectious diseases. Curr Proteomics 17:354–364. https://doi.org/10.2174/1570164617666190919120140
Gramatniece A, Silamikelis I, Zahare I et al (2019) Control of Acinetobacter baumannii outbreak in the neonatal intensive care unit in Latvia: whole-genome sequencing powered investigation and closure of the ward. Antimicrob Resist Infect Control 8:1–8. https://doi.org/10.1186/s13756-019-0537-z
Grote A, Hiller K, Scheer M et al (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. https://doi.org/10.1093/nar/gki376
Hajighahramani N, Nezafat N, Eslami M et al (2017) Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect Genet Evol 48:83–94. https://doi.org/10.1016/j.meegid.2016.12.010
Hansson T, Oostenbrink C, Van Gunsteren WF (2002) Molecular dynamics simulations. Curr Opin Struct Biol 12:190–196. https://doi.org/10.1016/S0959-440X(02)00308-1
Hoffman W, Lakkis FG, Chalasani G (2016) B cells, antibodies, and more. Clin J Am Soc Nephrol 11:137–154. https://doi.org/10.2215/CJN.09430915
Howard A, O’Donoghue M, Feeney A, Sleator RD (2012) Acinetobacter baumannii: an emerging opportunistic pathogen. Virulence 3:5. https://doi.org/10.4161/viru.19700
Ito T, Connett JM, Kunkel SL, Matsukawa A (2013) The linkage of innate and adaptive immune response during granulomatous development. Front Immunol 4:10. https://doi.org/10.3389/fimmu.2013.00010
Jamroz M, Kolinski A, Kmiecik S (2013) CABS-flex: server for fast simulation of protein structure fluctuations. Nucleic Acids Res 41:W427–W431. https://doi.org/10.1093/nar/gkt332
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105:6474–6487. https://doi.org/10.1021/jp003919d
Kar T, Narsaria U, Basak S et al (2020) A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep 10:1–24. https://doi.org/10.1038/s41598-020-67749-1
Karageorgopoulos DE, Falagas ME (2008) Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect Dis 8:751–762. https://doi.org/10.1016/S1473-3099(08)70279-2
Kim CH, Jeong YJ, Lee J et al (2013) Essential role of toll-like receptor 4 in Acinetobacter baumannii-induced immune responses in immune cells. Microb Pathog 54:20–25. https://doi.org/10.1016/j.micpath.2012.08.008
Kozakov D, Hall DR, Xia B et al (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12:255–278. https://doi.org/10.1038/nprot.2016.169
Kumar Pandey R, Ojha R, Mishra A, Kumar Prajapati V (2018) Designing B- and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J Cell Biochem 119:7631–7642. https://doi.org/10.1002/jcb.27110
Larsen MV, Lundegaard C, Lamberth K et al (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8:1–12. https://doi.org/10.1186/1471-2105-8-424
Liang Z, Zhu H, Wang X et al (2020) Adjuvants for coronavirus vaccines. Front Immunol 11:2896. https://doi.org/10.3389/fimmu.2020.589833
Livingston B, Crimi C, Newman M et al (2002) A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 168:5499–5506. https://doi.org/10.4049/jimmunol.168.11.5499
Manchanda V, Sinha S, Singh N (2010) Multidrug resistant Acinetobacter. J Glob Infect Dis 2:291. https://doi.org/10.4103/0974-777x.68538
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. https://doi.org/10.1093/bioinformatics/16.4.404
Moriel DG, Beatson SA, Wurpel DJ et al (2013) Identification of novel vaccine candidates against multidrug-resistant Acinetobacter baumannii. PLoS ONE 8:e77631. https://doi.org/10.1371/journal.pone.0077631
Morris FC, Dexter C, Kostoulias X et al (2019) The mechanisms of disease caused by Acinetobacter baumannii. Front Microbiol 10:1601. https://doi.org/10.3389/fmicb.2019.01601
Nain Z, Abdulla F, Rahman MM et al (2020) Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J Biomol Struct Dyn 38:4850–4867. https://doi.org/10.1080/07391102.2019.1692072
Nezafat N, Karimi Z, Eslami M et al (2016) Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput Biol Chem 62:82–95. https://doi.org/10.1016/j.compbiolchem.2016.04.006
Nie D, Hu Y, Chen Z et al (2020) Outer membrane protein A (OmpA) as a potential therapeutic target for Acinetobacter baumannii infection. J Biomed Sci 27:1–8. https://doi.org/10.1186/s12929-020-0617-7
Nosrati M, Hajizade A, Nazarian S et al (2019) Designing a multi-epitope vaccine for cross-protection against Shigella spp: an immunoinformatics and structural vaccinology study. Mol Immunol 116:106–116. https://doi.org/10.1016/j.molimm.2019.09.018
Noto MJ, Boyd KL, Burns WJ et al (2015) Toll-like receptor 9 contributes to defense against Acinetobacter baumannii infection. Infect Immun 83:4134–4141. https://doi.org/10.1128/IAI.00410-15
Oli AN, Obialor WO, Ifeanyichukwu MO et al (2020) Immunoinformatics and vaccine development: an overview. ImmunoTargets Ther 9:13–30. https://doi.org/10.2147/itt.s241064
Patra P, Bhattacharya M, Sharma AR et al (2020) Identification and design of a next-generation multi epitopes bases peptide vaccine candidate against prostate cancer: an in silico approach. Cell Biochem Biophys 2020 784 78:495–509. https://doi.org/10.1007/S12013-020-00912-7
Paul S, Weiskopf D, Angelo MA et al (2013) HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol 191:5831–5839. https://doi.org/10.4049/jimmunol.1302101
Ponomarenko J, Bui HH, Li W et al (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9:1–8. https://doi.org/10.1186/1471-2105-9-514
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5:9862. https://doi.org/10.1371/journal.pone.0009862
Rémy V, Zöllner Y, Heckmann U (2015) Vaccination: the cornerstone of an efficient healthcare system. J Mark Access Heal Policy 3:27041. https://doi.org/10.3402/jmahp.v3.27041
Ren S, Guan L, Dong Y et al (2019) Design and evaluation of a multi-epitope assembly peptide vaccine against Acinetobacter baumannii infection in mice. Swiss Med Wkly. https://doi.org/10.4414/smw.2019.20052
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. https://doi.org/10.1038/nprot.2010.5
Russo TA, Beanan JM, Olson R et al (2013) The K1 capsular polysaccharide from Acinetobacter baumannii is a potential therapeutic target via passive immunization. Infect Immun 81:915–922. https://doi.org/10.1128/IAI.01184-12
Saadi M, Karkhah A, Nouri HR (2017) Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect Genet Evol 51:227–234. https://doi.org/10.1016/j.meegid.2017.04.009
Samad A, Ahammad F, Nain Z et al (2020) Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1792347
Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA (2017) Fundamentals and methods for T- and B-cell epitope prediction. J Immunol Res. https://doi.org/10.1155/2017/2680160
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33:W363–W367. https://doi.org/10.1093/NAR/GKI481
Scott P, Deye G, Srinivasan A et al (2007) An outbreak of multidrug-resistant Acinetobacter baumannii-calcoaceticus complex infection in the US military health care system associated with military operations in Iraq. Clin Infect Dis 44:1577–1584. https://doi.org/10.1086/518170
Sefid F, Rasooli I, Jahangiri A, Bazmara H (2015) Functional exposed amino acids of BauA as potential immunogen against Acinetobacter baumannii. Acta Biotheor 63:129–149. https://doi.org/10.1007/s10441-015-9251-2
Sette A, Livingston B, McKinney D et al (2001) The development of multi-epitope vaccines: epitope identification, vaccine design and clinical evaluation. Biologicals 29:271–276. https://doi.org/10.1006/biol.2001.0297
Shey RA, Ghogomu SM, Esoh KK et al (2019) In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. https://doi.org/10.1038/s41598-019-40833-x
Singh R, Garg N, Shukla G et al (2016) Immunoprotective efficacy of Acinetobacter baumannii outer membrane protein, FilF, predicted in silico as a potential vaccine candidate. Front Microbiol 7:158. https://doi.org/10.3389/fmicb.2016.00158
Singh R, Capalash N, Sharma P (2017) Immunoprotective potential of BamA, the outer membrane protein assembly factor, against MDR Acinetobacter baumannii. Sci Rep 7:1–11. https://doi.org/10.1038/s41598-017-12789-3
Solanki V, Tiwari M, Tiwari V (2019) Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci Rep 9:1–19. https://doi.org/10.1038/s41598-019-41496-4
Song X, Zhang H, Zhang D et al (2018) Bioinformatics analysis and epitope screening of a potential vaccine antigen TolB from Acinetobacter baumannii outer membrane protein. Infect Genet Evol 62:73–79. https://doi.org/10.1016/j.meegid.2018.04.019
Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S (2015) An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform 53:405–414. https://doi.org/10.1016/j.jbi.2014.11.003
Tahir ul Qamar M, Shokat Z, Muneer I et al (2020) Multiepitope-based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines 8:288. https://doi.org/10.3390/vaccines8020288
Tomar N, De RK (2010) Immunoinformatics: an integrated scenario. Immunology 131:153–168. https://doi.org/10.1111/j.1365-2567.2010.03330.x
Vajda S, Yueh C, Beglov D et al (2017) New additions to the ClusPro server motivated by CAPRI. Proteins Struct Funct Bioinforma 85:435–444. https://doi.org/10.1002/prot.25219
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:407–410. https://doi.org/10.1093/nar/gkm290
Xie G, Gao S, Ou J et al (2021) Conjugating peptides onto 1D Rodlike bionanoparticles for enhanced activity against gram-negative bacteria. Nano Lett 21:1722–1728. https://doi.org/10.1021/ACS.NANOLETT.0C04516
Xu D, Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101:2525–2534. https://doi.org/10.1016/j.bpj.2011.10.024
Yang Z, Bogdan P, Nazarian S (2021) An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Reports 2021 111 11:1–21. https://doi.org/10.1038/s41598-021-81749-9
Zadeh Hosseingholi E, Rasooli I, Mousavi Gargari SL (2014) In silico analysis of Acinetobacter baumannii phospholipase D as a subunit vaccine candidate. Acta Biotheor 62:455–478. https://doi.org/10.1007/s10441-014-9226-8
Zhang Y, Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins Struct Funct Genet 57:702–710. https://doi.org/10.1002/PROT.20264
Zhou WY, Shi Y, Wu C et al (2009) Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine 27:5013–5019. https://doi.org/10.1016/j.vaccine.2009.05.009
Acknowledgements
The authors would like to acknowledge the computational help to run molecular dynamics simulations by Lab Engineer Muhammad Hassan Khan from Research Center for Modeling and Simulation (RCMS), National University of Sciences and Technology (NUST), Islamabad, Pakistan.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
KK designed and performed experiments and wrote the main manuscript text, SI, performed data curation, phylogenetic analysis and helped in the writing, SRU, helped in data analysis, immune simulations experiment and writing and SA devised and supervised experiments, analyzed data, and helped in writing the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khalid, K., Irum, S., Ullah, S.R. et al. In-Silico Vaccine Design Based on a Novel Vaccine Candidate Against Infections Caused by Acinetobacter baumannii. Int J Pept Res Ther 28, 16 (2022). https://doi.org/10.1007/s10989-021-10316-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-021-10316-7