
Monoclonal antibodies have been successfully utilized as cancer-targeting therapeutics and diagnostics, but the efficacies of these treatments are limited in part by the size of the molecules and non-specific uptake by the reticuloendothelial system. Peptides are much smaller molecules that can specifically target cancer cells and as such may alleviate complications with antibody therapy. Although many endogenous and exogenous peptides have been developed into clinical therapeutics, only a subset of these consists of cancer-targeting peptides. Combinatorial biological libraries such as bacteriophage-displayed peptide libraries are a resource of potential ligands for various cancer-related molecular targets. Target-binding peptides can be affinity selected from complex mixtures of billions of displayed peptides on phage and further enriched through the biopanning process. Various cancer-specific ligands have been isolated by in vitro, in vivo, and ex vivo screening methods. As several peptides derived from phage-displayed peptide library screenings have been developed into therapeutics in current clinical trials, which validates peptide-targeting potential, the use of phage display to identify cancer-targeting therapeutics should be further exploited.
Similar content being viewed by others

Introduction
Antibody engineering has provided many advances in tumor-targeted therapeutics and diagnostics. As recombinant monoclonal antibodies (mAbs) posses the general properties of high target specificity and affinity, they are well-suited therapeutic agents and greater than 400 mAbs are currently in clinical trials (Lin et al., 2005). Current Food and Drug Administration approved antibody therapeutics in clinical use include rituximab (anti-CD20 mAb against Non-Hodgkin lymphoma, and B-cell lymphomas) and its related radioimmunoconjugates; gemtuzumab ozogamicin (anti-CD33 mAb conjugated to a cytotoxic agent against acute myeloid leukemia); alemtuzumab (anti-CD52 mAb against B-cell chronic lymphocytic leukemia); cetuximab (anti-epidermal growth factor receptor mAb against colorectal cancer); trastuzumab (anti-HER2 mAb against breast cancer); and bevacizumab (anti-vascular endothelial growth factor mAb against metastatic colorectal cancer) (Adams and Weiner, 2005; Levene et al., 2005). The successes of these therapeutic antibodies validate the principle that cancer can be treated through a cell surface targeted approach (Aina et al., 2005).
Although antibody-based therapeutics have increased the outlook for many cancer patients, these therapies are of limited efficacy especially against solid malignancies (Stern and Herrmann, 2005) due in part to the size of the molecules (approximately 160 kDa), elevated tumor interstitial pressure, non-specific uptake into the reticuloendothelial system, and immunogenicity (Jiang et al., 2004; Aina et al., 2005). In addition, high affinity targeting antibodies experience a ‘binding site barrier’ that limits tumor perfusion (Adams et al., 2001). Engineering antibodies into Fab, and single-chain Fv fragments (approximately 25 kDa), and VHH domains or ‘nanobodies’ (approximately 15 kDa), has offered improvements in tumor penetration and in decreased immunogenicity (Cortez-Retamozo et al., 2004).
Research indicates that pharmacokinetic properties are improved with the use of smaller ligands (Reilly et al., 1995). As peptides are much smaller molecules (1–2 kDa) than antibodies or antibody fragments and demonstrate specificity and affinity for their targets, they may in part alleviate the complications associated with antibody therapy (Ladner et al., 2004). In general, peptides demonstrate better tumor penetration, are subject to less non-specific uptake, and should not elicit an immune response. In addition, although therapeutic peptides are vulnerable to proteolytic degradation, for enhanced affinity and stability, peptides can be engineered with d-amino acids, cyclized, and blocked at the N- and C-termini (Nilsson et al., 2000; Aina et al., 2002).
Various endogenous peptides regulate bodily functions such as peptide hormones, chemoattractants, neurotransmitters, cytotoxins, and antimicrobials. The discovery of endogenous peptides led to the development of these peptides as targeting therapeutics. For example, somatostatin, a naturally occurring inhibitory peptide, was discovered to have anti-neoplastic effects on cells expressing somatostatin receptors. A somatostatin analogue octreotide has been utilized to treat a variety of tumors. It has been conjugated to radiochemicals for diagnostic imaging and to radionuclides for targeted chemotherapeutic delivery (Liu, 2005). Recently discovered bioactive endogenous peptides include hepcidin, LEAP-2, β and θ-defensins, various ligands of orphan G-protein-coupled receptors, and lympho-epithelial Kazal-type serine protease inhibiting peptides (Adermann et al., 2004). Although it is estimated that greater than 850 endogenous peptides exist, only a small subset of these consists of cancer-associated peptides.
In addition to the clinical use of endogenous peptides and their analogs, several exogenous peptides have also been developed into clinical therapeutics. Enfuvirtide, a 36-amino acid synthetic peptide derived from human immunodeficiency virus (HIV)-1 glycoprotein gp41, was the first marketed viral fusion inhibitor for the treatment of HIV infection (Matthews et al., 2004). Ziconotide, a synthetic equivalent of a naturally occurring 25-amino acid peptide derived from the marine snail Conus magnus, has been approved for treating acute and chronic pain (Miljanich, 2004; Winquist et al., 2005). Exenatide, a 39-amino acid peptide isolated from the gila monster Heloderma suspectum, is an incretin mimetic used to enhance glucose-dependent insulin secretion in patients with type 2 diabetes mellitus (Nielsen et al., 2004; Roges et al., 2005).
Phage-displayed random peptide libraries are a valuable screening resource for identifying bioactive peptides that interact with cancer targets (Mori, 2004). A typical library contains as many as 109 peptides, and often, multiple binding peptides can be identified during peptide library screenings. Peptide therapeutics developed through phage-display technology currently in clinical trials include: DX-88, an inhibitor of plasma killikrein for the treatment of hereditary angioedema (Ritchie, 2003); DX-890, an inhibitor of human neutrophil elastase for treatment of cystic fibrosis (Wark, 2002); and Hematide, a synthetic peptide-based erythropoiesis-stimulating agent.*Footnote 1 A number of papers have been published describing the isolation of tumor and/or tumor vasculature-targeting peptides from phage-displayed peptide libraries (Landon and Deutscher, 2003; Zurita et al., 2003; Haubner and Wester, 2004; Ruoslahti, 2004; Shadidi and Sioud, 2004; Kelly et al., 2005; Su et al., 2005). This review attempts to provide the reader an update on the status of cancer-targeted peptides isolated from phage-displayed peptide libraries.
Phage-Displayed Peptide Library
Background
The field of phage display began in 1985 when George Smith reported that filamentous bacteriophage (phage) could be manipulated to display foreign amino acid sequences on the surface of the phage particle as phage coat protein covalent fusions, while maintaining the ability of phage infectivity for propagation (Smith, 1985). Combinatorial phage-displayed peptide libraries have since emerged as a screening resource for identifying peptide ligands of molecular targets. Cancer-specific ligands have been isolated by in vitro, in vivo, and ex vivo library screening methods. The reader is referred to a review article covering identified peptidic ligands (Mori, 2004). The technology has also been applied in various other applications, such as studying protein-ligand interactions, affinity maturation of previously isolated binding peptides, mapping epitope binding sites, and identifying enzyme substrate specificity (Azzazy and Highsmith, 2002).
The utility of phage-displayed peptide library technology derives from phage biology. Phage are DNA-containing viruses that infect bacteria. Phage can be genetically engineered to display polypeptides on their surfaces that are encoded by the DNA encapsulated in the viral particle. As such, phage phenotype is physically linked to phage genotype. Libraries are created with standard molecular cloning techniques, which involve insertion of randomized oligonucleotide fragments in frame into the phage coat protein genes. Phage displaying peptides that interact with a molecular target can be affinity selected from complex mixtures of billions of displayed peptides, propagated by in vivo amplification, and subjected to additional rounds of affinity selection, the process of which is referred to as “biopanning.”
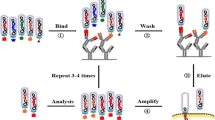
A typical round of biopanning includes (1) incubating the peptide library with the target, (2) washing away unbound phage, (3) eluting the remaining bound phage, and (4) amplifying the eluted phage for subsequent screening rounds (Fig. 1). After multiple rounds of selection, enrichment of target-binding phage is detected by phage titering and/or immunological assay methods. When sufficient enrichment has occurred, individual phage are isolated and sequenced to reveal any enriched binding motif. Each step of the biopanning process can be optimized to tailor to the unique characteristics of the target, and the desired result of the screening project. For instance, purified target proteins can be presented in various formats, such as indirect immunoadhesion formats, or captured through an affinity tag (Mori, 2004). The criterion for target protein presentation in a phage-displayed peptide library screening is that the protein approximates its native conformation, as evidenced by the retention of biological activity, such as enzymatic activity or ability to participate in protein–protein interactions. Screening projects can also be designed to select for high affinity binding peptides by increasing the stringency of phage binding, washing, and/or elution steps and by imposing partial structure of the displayed peptides by specifying amino acid residues during library construction. Typical dissociation constants of isolated peptides are in the low to mid-micromolar range (Kay et al., 2001).

Affinity selection (biopanning) process employed in phage-displayed peptide library screenings. The naïve peptide library is incubated with the target, washed to remove non-specific binding phage, and eluted to collect binding phage. The eluted phage are then amplified in vivo and subjected to additional rounds of biopanning, or can be plated to isolate clones for sequencing.
Limits of Phage-Displayed Peptide Library Technology
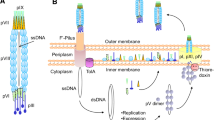
Two types of Escherichia coli bacteriophages have been used in peptide phage-display systems, filamentous phage (M13, f1, and fd) and lytic phage (T7, T4, and λ) (Castagnoli et al., 2001). Filamentous phage particles are approximately 5 nm in diameter and 1 μm in length and consist of major and minor coat proteins and circular single-stranded DNA (Fig. 2a). Peptides are typically displayed as N-terminal gene III protein (pIII) fusions or gene VIII protein (pVIII) fusions. pIII is a minor coat protein, present in five copies at one tip of the phage particle. Peptides fused to this protein are also expressed in low copy numbers or monovalent display with the use of a phagemid vector. pVIII is the M13 major coat protein, of which thousands of copies encapsulate the phage genome. Peptides fused to this protein can be expressed in high copy number, which is sometimes referred to as landscape phage display (Petrenko and Smith, 2000), or low to mid-copy number using a phagemid system. For the display of peptides as fusions to each coat protein molecule present in the phage particle, the randomized DNA is inserted directly into the phage coat protein gene. Conversely, for peptide displayed on only a few of the coat protein molecules, a phagemid vector supplying the randomized DNA is utilized in conjunction with helper phage infection (Russel et al., 2004). On the other hand, bacteriophage T7 consists of an approximately 60 nm diameter head encapsulating linear double-stranded DNA, a tail, and six tail fibers (Fig. 2b). Peptides are displayed on the head surface of the T7 particle as C-terminal fusions to capsid protein 10. There are six T7 cloning vectors available for use in the T7Select system (Novagen) that accommodate amino acid lengths ranging from 50 to 1200, in peptide copy numbers of 0.1–1, 5–15, or 415*Footnote 2 depending on vector selection (Rosenberg et al., 1996).

(a) Structure of a typical filamentous phage virion, and (b) a T7 lytic phage virion.
A critical aspect of peptide phage-display technology is the complexity of the library used during screening, as successful isolation of peptides for a target requires the presence of the peptide in the library chosen for biopanning. That is, the library should be of sufficient diversity to contain potential binding ligands for the target. Most phage-display peptide library screenings have been conducted using filamentous phage-displayed peptides (Szardenings, 2003), even though previous studies demonstrated specific and positional amino acid biases in populations of peptides randomly selected from M13 libraries (Rodi et al., 2002). Although detailed phage biology is beyond the scope of this review article, for careful consideration of this aspect, the phage morphogenesis process within the biological system chosen for peptide display should be considered.
Phage morphogenesis can be divided into three distinct steps: infection of the host cell, translation and assembly of the phage particles, and release to the extracellular environment. Each of these steps has the potential to impose amino acid sequence bias on a phage-displayed peptide library (Rodi and Makowski, 1999). Filamentous phages initiate the infection process by an interaction between pIII at the tip of the phage particle and E. coli F pili, assemble through a complex inner-membrane associated process involving bacterial and phage proteins and are then secreted through the host outer membrane (Fig. 3a) (Russel et al., 2004). On the other hand, lytic phages initiate infection through an interaction between their tail fibers and lipopolysaccharide on the E. coli cell surface, after which the phage genome and several proteins are injected into the host cell through its tail (Kemp et al., 2005). Lytic phage assembly occurs within the E. coli cytoplasm, and mature phage virions are released by cell lysis (Fig. 3b). For more information regarding phage morphogenesis, the reader is referred to the following phage-display manuals (Barbas et al., 2001; Clackson and Lowman, 2004).

Differing processes of filamentous phage and lytic phage morphogenesis. (a) Filamentous phage assembles at the E. coli inner-membrane (IM) and is secreted through the outer-membrane (OM) into the extracellular environment, a process that preserves host viability. (b) Lytic phage assembles within the E. coli cytoplasm, and mature virions are released by cell lysis.
Because peptides displayed on lytic phage do not have to be compatible with the host cell synthesis and secretion apparatuses, libraries produced with lytic phage can surpass the diversity of filamentous phage-displayed peptide libraries. A recent bioinformatics-assisted comparison of T7 lytic phage and M13 filamentous phage-displayed peptide libraries in our laboratory demonstrated that peptide libraries produced with the T7 system have less amino acid biases than libraries produced with the M13 system (Table I). The overall peptide diversity estimate for a T7 12-mer random library (T7 12-mer NNK) demonstrated a 14-fold increase over the peptide diversity estimate for an M13 12-mer random library (M13 12-mer NNK). For this comparison, the T7 and M13 libraries analyzed were constructed using the reduced genetic code method of library oligonucleotide DNA design, or ‘NNK’ method (see below for further discussion).
New Construction Methods of Phage-Displayed Peptide Library Increase Diversity
Another factor contributing to the diversity of a phage-displayed peptide library is the method of library construction. When designing peptide-encoding oligonucleotide DNA to be inserted into the phage genome, the type of genetic code employed is of utmost importance because the observed frequency of amino acids displayed in the library is directly correlated to the number of codons encoding each amino acid. For instance, the standard 64-codon genetic code encodes each of the twenty amino acids and three stop codons with the number of codons per amino acid ranging from one (methionine, tryptophan) to six (leucine, serine, arginine). Amino acids encoded by higher numbers of codons have a greater chance of being incorporated into the library, while amino acids with fewer codons have a lesser chance. This discrepancy results in non-uniform amino acid frequencies and thus, limited peptide diversity.
Ideally, a library would contain peptides with even 5% amino acid frequencies (one amino acid out of twenty) per each amino acid position within the peptides. To produce libraries with more even amino acid frequencies, phage-displayed peptide libraries are typically created using a reduced genetic code when designing the insert oligonucleotide DNA encoding the displayed peptides. The oligonucleotides are designed in the ‘NNK’ format, where N represents equal proportions of guanine, cytosine, thymine, and adenine nucleotides, and K represents equal proportions of thymine and guanine nucleotides. This method encodes all twenty amino acids and one stop codon, while smoothing the number of codons per amino acid to one, two, or three (Scott, 2001). Although the NNK method of library construction provides each amino acid a more equal opportunity of incorporation into the peptide library, the method still imparts some amino acid sequence bias because the amino acids are encoded by varying numbers codons.
An alternative method of library construction that alleviates the biases imposed by randomized oligonucleotide DNA methods involves using codon-corrected trinucleotide cassettes. In this method, codons for each amino acid are first synthesized as individual trimer nucleotide cassettes, which are then mixed in appropriate ratios according to the codon reaction efficiencies to form the peptide-encoding DNA to be inserted into the phage genome. This method offers several advantages over the randomized nucleotide method of library construction. Undesired stop codons are eliminated from insert DNA, and because each amino acid is encoded by only one codon, the method allows for a predefined ratio of amino acids to be incorporated at each synthesis step. This method is also advantageous in that it allows the user to define which amino acid codons to use based on E. coli codon usage. Previous research indicates that atypical amino acid codon usage stresses the host translation system (Kayushin et al., 1996; Rodi and Makowski, 1999), which may result in the loss of peptides containing amino acids encoded by rare codons, and thus limit overall peptide diversity. Recent research in our laboratory has shown that a random 12-mer peptide library constructed with the T7 lytic phage system in combination with the trinucleotide cassettes (Kayushin et al., 1996) resulted in increased peptide diversity, as compared to libraries constructed with M13 filamentous phage system and the NNK method (Table I). The analysis revealed a 3-fold increase in peptide diversity when using trinucleotide cassettes (T7 12-mer tri-nucleotide) as compared to the NNK method (T7 12-mer NNK), and a 43-fold increase in peptide diversity when using T7 lytic phage and trinucleotide cassettes, as compared to using M13 filamentous phage and the NNK method (M13 12-mer NNK).
Tumor-Targeting Peptides Isolated from Phage-Displayed Peptide Libraries
Within the last several years, there were a number of articles describing tumor-targeting peptides isolated from phage-displayed peptide library screenings. In these research papers, filamentous phage-display systems, such as M13 phage, were used in most publications, and only a handful groups used lytic phage-displayed systems, such as T7 phage. Approximately 50% of these articles used linear peptide libraries, and constrained peptide libraries were used in the other half of the publications. With regard to the formats of targets used in biopanning, about 40% of these articles used recombinantly expressed proteins that were over-expressed or specifically expressed in and on tumor cells. Tumor cell lines and freshly excised tumor cells were used as targets in 50% of these research articles without knowing the exact identity of the target molecules on tumor cells. The last 10% of these publications used in vivo and/or ex vivo biopanning methods. All these research publications showed that specificity of the isolated phage clones and/or corresponding peptides bound to the target molecules in enzyme-linked immunosorbent assay formats as well as to target cells in immunoblotting, immunoprecipitation, cell-binding assays, flow cytometric analyses or immunostaining of cells and tissues. Many of the isolated peptides, which bound to the targets, were internalized into the cells. However, most studies did not evaluate the targeting efficiency and specificity of isolated peptides in vivo. In this article, we decided to review only the articles within the last several years in which peptides isolated from phage-displayed peptide libraries were evaluated in vivo. The reader is referred to the recent review articles covering the subject (Aina et al., 2002; Romanov, 2003; Mori, 2004).
Screening Against Recombinant Tumor-Associated Proteins
Screening phage-displayed peptide libraries against homogenous recombinant tumor-associated proteins was the most direct and successful method of identifying peptidic ligands of target molecules. Portions of recombinant tumor membrane proteins including functionally folded extracellular domains, with and without immunoglobulin domains, were used during library screenings.
By screening a 9-mer random cyclic peptide filamentous phage library, the proapoptotic peptide P15, which targeted casein kinase 2 (CK2) phosphorylation site, was isolated (Perea et al., 2004). P15 peptide abrogated CK2 phosphorylation by blocking the substrate in vitro. P15 fused to a cell-penetrating peptide derived from the HIV-Tat protein (P15-Tat) induced apoptosis by rapid caspase activation and cellular cytotoxicity in a variety of tumor cell lines. In addition, in vivo administration of P15-Tat into C57BL6 mice bearing day 7-established solid tumors resulted in substantial regression of the tumor mass. This work suggested that the P15 cyclic peptide might potentially be used in the treatment of solid tumors.
A novel peptide S7, which selectively bound to interleukin-6 receptor (IL-6R) chain, was identified by screening an M13 phage-displayed 7-mer random cyclic peptide library (Su et al., 2005). The chemically synthesized S7 peptide blocked the interaction between interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) in a concentration-dependent manner, prevented IL-6-mediated survival signaling, and sensitized cervical cancer cells to chemotherapeutic compounds in vitro. It was revealed that the S7 peptide substantially inhibited IL-6-induced vascular endothelial growth factor-A expression and angiogenesis in different cancer cell lines. In addition, the authors showed that S7 peptide significantly suppressed IL-6-induced vascular endothelial growth factor-mediated cervical tumor growth in severe combined immunodeficient mice. These observations highlighted the potential use of a small peptide, which blocks IL-6/IL-6R interaction, in the management of patients with malignant disease.
Screening Against Whole Cells in Cell Culture
Although peptidic ligands for recombinant tumor membrane proteins can be identified using phage-displayed peptide library technology, the peptides may not have access to their target in vivo due to post-translational modifications and malignancy-specific modifications. In addition, it is often difficult to recombinantly produce soluble and functionally folded extracellular domains of membrane proteins. These issues have prompted many investigators to use whole intact cells to screen phage-displayed peptide libraries in vitro. Using this method of biopanning, specific peptidic ligands for cell surface molecules and cell-internalizing peptides were identified.
An M13 phage-displayed 12-mer random peptide library was screened to isolate phage that bound specifically to human glioma cell lines (Ho et al., 2004). One of the isolated peptides, MG11, was glioma-specific and gave an in vitro binding enrichment of more than 5-fold for glioma cells when compared with non-glioma cells. Intravenous injection of phages displaying the MG11 peptide enabled the phage to target specifically to glioma xenografts. Moreover, when Lissamine rhodamine-labeled MG11 peptide was administered intratumorally, it targeted specifically to glioma xenografts.
Screening of an M13 phage-displayed 7-mer random cyclic peptide library was performed on murine endothelium under physiological flow conditions to isolate a family of endothelial vascular adhesion molecule-1 (VCAM-1)-mediated cell-internalizing peptides (Kelly et al., 2005). One specific sequence (VHSPNKK) having homology to the alpha-chain of very late antigen (a known ligand for VCAM-1) bound VCAM-1 and interfered with leukocyte-endothelial interactions. The peptide showed 12-fold higher target-to-background ratios as compared with a VCAM-1 monoclonal antibody. A VHSPNKK-modified magnetofluorescent nanoparticle (VNP) demonstrated high affinity for VCAM-1-expressing endothelial cells but very low affinity for macrophages. VNP targeted VCAM-1-expressing endothelial cells in a murine tumor necrosis factor-induced inflammatory in vivo model and co-localized with VCAM-1-expressing cells in atherosclerotic lesions present in cholesterol-fed apolipoprotein E apoE-/- mice. These results indicated that the VNP could be useful for in vivo imaging of endothelial markers by MRI and fluorescence imaging.
A peptide PA1 that internalized to irradiated Capan-2 pancreatic adenocarcinoma cells was isolated from an M13 phage-displayed 12-mer random linear peptide library (Huang et al., 2005). Fluorescein-labeled PA1 peptide was able to penetrate tumor tissue after intravenous injections to Capan-2 xenografts in nude mice and bound specifically to irradiated tumor cells.
An M13 phage-displayed 12-mer random linear peptide library was screened against the prostate-specific membrane antigen-negative cell line DU-145 (Zitzmann et al., 2005). The isolated peptide DUP-1 was evaluated in vitro for its binding specificity, kinetics, affinity, and internalization of the peptide. Biodistribution studies of 131I-labeled DUP-1 in nude mice with subcutaneously injected DU-145 and PC-3 tumors showed accumulation of the peptide in the tumors. The rat prostate tumor model AT-1 also demonstrated an increase of radioactivity in the prostate tumor up to three-fold in comparison with normal prostate tissue.
In vivo and Ex vivo Screening
In vivo screening of phage-displayed peptide libraries involves injecting the library intravenously into a living host, collecting the tissue of interest, rescuing the phage clones from that tissue, and subjecting the recovered phage to additional rounds of selection. After completing several rounds of screening, the recovered phage clones are analyzed for binding-motifs. Using this method of phage-displayed peptide library screening offers advantages over other methods of selection in that phage are exposed to the target molecules in their native environment. Phage clones displaying peptides that interact with non-target molecules, such as ubiquitous cell surface proteins and plasma proteins, are depleted from the phage population. In addition, only peptides that survive the degradative environment of the vascular system and only those that can access their targets are selected and propagated (Kolonin et al., 2001). The identification of peptides that target selective vasculature through in vivo phage-displayed peptide library screenings was first reported by Pasqualini and Ruoslahti (1996) and has since been reviewed (Rafii et al., 2003; Zurita et al., 2003; Mori, 2004; Ruoslahti, 2004).
In 2002, the first in vivo screening of a phage-displayed peptide library in a human patient was reported (Arap et al., 2002b). After one round of screening, phage that homed to different organs were isolated, and it was found that peptide binding to these different cell types did not occur by a random process. To validate tissue specificity of a peptide, the group demonstrated that an isolated phage displaying the peptide interacted with the interleukin-11 receptor (IL-11R) present in prostate tissue. IL-11R was further validated as a candidate for molecularly targeted prostate cancer therapeutics (Zurita et al., 2004). After demonstrating minimal toxicity of the in vivo screening of phage-displayed peptide libraries in mice, another group received Food and Drug Administration approval for the use of the technique in human clinical trials (Krag et al., 2002).
As the result of an in vivo screening of filamentous phage-displayed random peptide libraries, peptides that specifically recognized the vasculature in the prostate were identified (Arap et al., 2002a). One of the phage clones displaying the peptide SMSIARL homed to the prostate 10–15 times more than to other organs, as well as bound to vasculature in the human prostate. Synthetic SMSIARL peptide inhibited the prostate-homing of SMSIARL-bearing phage when co-injected into mice. Systemic treatment of mice with the peptide linked to a proapoptotic peptide that disrupts mitochondrial membranes caused tissue destruction in the prostate. The development of the cancers in prostate cancer-prone transgenic mice (TRAMP mice) was also delayed by the chimeric peptide.
A cyclic peptide CPGPEGAGC that homes to normal breast tissue with a 100-fold selectivity over non-targeted phage was isolated after in vivo biopanning of a T7 phage-displayed 7-mer random cyclic peptide library (Essler and Ruoslahti, 2002). The CPGPEGAGC-bearing phage binds to the blood vessels in the breast, as well as the vasculature of hyperplastic and malignant lesions in transgenic breast cancer bearing mice. The authors also identified that the homing peptide bound to aminopeptidase P, which is widely expressed in malignant breast tissue.
Laakkonen et al. (2002) devised a phage screening procedure that would favor tumor-homing to targets that are accessible to circulating phage, but not blood vessels, by combining the ex vivo and in vivo screening procedures. For the ex vivo screening portion, tumor-cell suspensions were prepared from human MDA-MB-435 breast carcinoma xenograft tumors using collagenase to disperse the tissue. A T7 phage-displayed 7-mer random cyclic peptide library was first incubated with the cell suspension. The authors then used magnetic beads coated with anti-mouse CD31 to preferentially deplete the tumor-derived cell suspension of blood vessel endothelial cells. The ex vivo pre-selected phage pool that bound to the CD31-deficient cell population was then subjected to in vivo biopanning on MDA-MB-435 breast carcinoma xenografts. One of the peptide sequences enriched was CGNKRTRGC named LyP-1. The LyP-1-displaying phage bound to primary MDA-MB-435 tumor-derived cell suspensions about 7000 times more than non-recombinant phage. The fluorescein-labeled LyP-1 synthetic peptide was detected in tumor structures that were positive for three lymphatic endothelial markers and negative for three blood vessel markers and that were accumulated in the nuclei of the putative lymphatic cells and tumor cells. LyP-1 peptide also homed to an osteosarcoma xenograft and spontaneous prostate and breast cancers in transgenic mice.
Another group screened out novel peptides homing to angiogenic vessels formed by a dorsal air sac method from a filamentous phage-displayed random 15-mer linear peptide library (Oku et al., 2002). After the determination of the epitope sequences of some of the isolated peptides, a liposome was modified with the epitope penta-peptide APRPG. The liposome demonstrated high accumulation in murine tumor xenografts, and APRPG-modified liposome encapsulating adriamycin effectively suppressed experimental tumor growth. In addition, specific binding of APRPG-modified liposome to human umbilical endothelial cells was shown.
To identify homing peptides for blood vessels in a mouse model of HPV16-induced epidermal carcinogenesis, Hoffman et al. (2003) used a T7 phage-displayed 7-mer random cyclic peptide library for ex vivo/in vivo biopanning. One peptide, CSRPRRSEC, recognized the neovasculature in dysplastic skin but not in carcinomas. Two other peptides (CGKRK and CDTRL) preferentially targeted to neovasculature in tumors and to premalignant dysplasias to a lesser degree. These peptides did not home to vessels in normal skin, other normal organs, or the stages of pancreatic islet carcinogenesis in another mouse model.
In a combination of ex vivo and in vivo biopanning with a T7 phage-displayed random 7-mer cyclic peptide library, Joyce et al. (2003) profiled the vasculature in the angiogenic stages of a mouse model of pancreatic islet carcinogenesis. Seven homing peptides to angiogenic progenitors, solid tumors, or both were characterized. Five of them selectively targeted to neoplastic lesions in the pancreas and not to islet cell tumors growing subcutaneously, xenotransplant tumors from a human cancer cell line, or an endogenously arising squamous cell tumor of the skin. Three peptides with unique homing to angiogenic islets, tumors, or both co-localized with markers of endothelial cells or pericytes. One peptide was homologous with pro-PDGF-B expressed in endothelial cells.
In vivo biopanning with laser pressure catapult microdissection was utilized to screen for peptide bound vascular receptors in the islets of Langerhans in the murine pancreas from a filamentous phage-displayed 7-mer random cyclic peptide library (Yao et al., 2005). Two of the isolated peptides showed sequence identity to ephrin A-type ligand homologues. Confocal microscopy verified that most immunoreactivity of the phages displaying these two peptides was linked with blood vessels in pancreatic islets. Binding of both islet-homing phage and antibodies recognizing EphA4, a receptor for ephrin-A ligands, was dramatically escalated in blood vessels of pancreatic islet tumors in RIP-Tag2 transgenic mice, indicating that endothelial cells of blood vessels in pancreatic islets preferentially express EphA4 receptors, and this expression is increased in tumors.
Applications of Peptides Isolated from Phage-Displayed Peptide Libraries
Peptides isolated from phage-displayed libraries can be used as targeting molecules for many applications: peptides themselves, radiolabeled peptides, peptides conjugated with chemotherapeutic agents, peptides fused with toxins, and peptides on nanoparticles or liposomes carrying chemotherapeutic agents.
Antitumor Activity
Fluorescein-conjugated LyP-1, a peptide selected from a phage-displayed peptide library that specifically binds to tumor and endothelial cells of tumor lymphatics in certain tumors, strongly and specifically accumulated in primary MDA-MB-435 breast cancer xenografts and their metastases from intravenous peptide injections (Laakkonen et al., 2004). The LyP-1 peptide accumulation and hypoxic areas in tumors overlapped. Systemic LyP-1 peptide administration of mice with breast cancer xenograft inhibited tumor growth and reduced the number of tumor lymphatic vessels. The authors suggested that this unexpected anti-tumor effect by the LyP-1 peptide could be a starting point for the new anti-tumor agent targeting to specific tumor lymphatics.
Drug Delivery
All peptides specific to tumor and tumor vasculature conjugated with toxic molecules could be potentially used as therapeutic agents against tumors.
In 1998, Ruoslahti’s group reported that peptides isolated from in vivo biopanning of phage-displayed peptide libraries specifically homed to tumor blood vessels (Arap et al., 1998). One motif RGD that selectively bound to αvβ3 and αvβ5 integrins, and another motif NGR that selectively homed to tumor vasculature. The receptor for the NGR containing peptides was later identified as aminopeptidase N (Pasqualini et al., 2000). These peptides coupled with the anticancer drug doxorubicin enhanced the efficacy of doxorubicin against human breast cancer xenografts in nude mice and reduced its toxicity.
Pasqualini and colleagues conjugated the peptide containing the RGD or NGR with a programmed cell death-inducing peptide sequence, KLAKLAKKLAKLAK. The homing domains guided the entire peptides to targeted cells, and the pro-apoptotic domain disrupted mitochondrial membranes after internalization of the peptide. These peptides composed of two functional domains were selectively toxic to angiogenic endothelial cells and demonstrated anti-cancer activity in mice (Ellerby et al., 1999).
Nanomaterial Delivery
Over the last few years, the development of nanostructures that detect and monitor cancer markers in vivo and that target therapeutic and imaging agents to cancer lesions and their microenvironment has attracted broad interest in medical research (Sullivan and Ferrari, 2004; Ferrari, 2005).
Several homing peptides originally isolated from in vivo screening of phage-displayed peptide libraries were used for in vivo targeting of semiconductor quantum dots (qdots) to specific vascular sites in mice (Akerman et al., 2002). The qdots coated with a GFE-1 lung-targeting peptide accumulated in the lungs of mice after intravenous injection. Two other peptides named F3 and LyP-1 specifically targeted the qdots to blood vessels or lymphatic vessels in tumors. The authors also show that addition of polyethylene glycol to the qdot coating reduced non-selective accumulation of the qdots in reticuloendothelial tissues.
Kontermann’s group screened out novel high-affinity cyclic RGD peptides from phage-displayed RGD motif libraries against both endothelial and melanoma cells (Holig et al., 2004). Using one of the high-affinity peptides RGD10, the authors generated novel lipopeptides composed of a lipid anchor, a short flexible spacer, and the peptide ligand conjugated to the spacer end. Incorporation of RGD10 lipopeptides into liposomes caused specific and efficient binding of the liposomes to integrin-expressing cells. Furthermore, in vivo experiments applying doxorubicin-loaded RGD10 liposomes in a C26 colon carcinoma mouse model demonstrated improved efficacy.
Imaging
Matrix metalloproteinases (MMPs) play an important role in cancer as well as in numerous other diseases. The cyclic decapeptide containing HWGF that was originally selected from a phage-displayed peptide library against MMP-2 and MMP-9 was radiolabed to evaluate the ability of this labeled peptide to monitor MMP-2 and MMP-9 activity, biodistribution, competition studies and plasma metabolites analyses in Lewis Lung cancer tumor bearing mice (Kuhnast et al., 2004). However, the radiolabeled peptide was not a suitable tracer for targeting of MMP-2 and MMP-9 in vivo because of its poor solubility and metabolic instability. Further modification of this peptide is necessary to improve metabolic stability and hydrophilicity for further evaluation of this peptide in vivo as a marker of gelatinase activity.
Gene Targeting
As a gene-targeting molecule, a peptide containing the NGR motif was inserted into the capsid of adeno-associated virus vectors. These recombinant viruses showed an altered tropism toward cells expressing the CD13 receptor, to which the NGR motif binds (Grifman et al., 2001). A similar study using the NGR motif was reported (Liu et al., 2000). The authors incorporated the NGR motif containing peptide into the envelope protein of Moloney murine leukemia virus, and the engineered viruses efficiently transduced human endothelial cells.
Perspectives
Many target-binding peptides isolated through the screening of phage-displayed random peptide libraries modulate the biological function of target molecules. Therefore, it is possible that certain tumor and tumor vasculature-targeting bioactive peptides can directly serve as agonist or antagonists. However, in general, peptides directly isolated form phage-displayed random peptides bind to their targets with low affinity (micromolar range). Thus, it would be necessary to increase the affinity of the peptides. One way to improve affinity is to screen second-generation peptide libraries displayed on phage based on core motifs identified from random peptide libraries (Deshayes et al., 2002; Fleming et al., 2005). Using this method, it is possible to increase the binding affinity of target-interacting peptides to the nanomolar range (100–1000-fold lower than that of first-generation peptides).
One major bottleneck of all biological display systems, including phage-displayed combinatorial peptide libraries, is the use of only the 20 natural l-amino acids that are prone to degradation by proteases. Thus, selected peptides are susceptible to proteolytic degradation in vivo unless (1) their amino and carboxyl termini are protected, (2) they are cyclized, and/or (3) they contain d-amino acids (Aina et al., 2002). However, it is now possible to genetically encode unnatural amino acids with diverse physical, chemical, or biological properties in E. coli, yeast, and mammalian cells (Xie and Schultz, 2005). In 2004, Schultz’s group first reported a phage display system with unnatural amino acids (Tian et al., 2004). This could allow not only the increase of the complexity of phage-displayed peptide libraries, but also the enhancement of the proteolytic stability of peptides isolated from phage display systems.
The other method to desensitize proteolytic degradation of peptides identified through phage-displayed peptide libraries is the mirror-image phage display approach (Wiesehan and Willbold, 2003). This technique was used by Kim’s group to identify d-peptides that bind to the SH3 domain of c-Src (Schumacher et al., 1996) as well as the coiled-coil pocket of HIV-1 gp41 (Eckert et al., 1999). Recently, Wiesehan et al. isolated d-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide A1-42 by the mirror-image phage display approach (Wiesehan et al., 2003). Only a few examples of this application were reported perhaps because it is expensive to synthesize d-peptides, and the size of a given target molecule should be small due to limitation of synthetically available peptide length.
Lam et al. (1991) first reported the “one-bead, one-compound” combinatorial libraries, which may contain l-amino acids, d-amino acids, unnatural amino acids, and even non-peptidic moieties. His group has applied the “one-bead, one-compound” combinatorial library method to successfully discover peptide ligands for a number of different human cancer cell lines (Aina et al., 2002). Perhaps, the combination of the phage-displayed peptide library technique and the “one-bead, one-compound” combinatorial library method should be explored to identify peptides with high affinity and resistance to proteolysis. A limitation of the “one-bead, one-compound” combinatorial library is the realistic size of the library (up to millions of compounds). It may be a good idea to screen phage-displayed random peptide libraries to identify target-binding motifs at first and subsequently screen second-generation “one-bead, one compound” combinatorial libraries in which the target-binding motifs are incorporated. This idea may facilitate the discovery and development of peptide/peptidomimetics that are directly useful as diagnostic and therapeutic agents.
All the peptides originally isolated from phage-displayed random peptide libraries could be used as leads for the rational design of peptidomimetic compounds (Kay et al., 1998; Nixon, 2002). The information obtained from structure-activity relationships and the conformational properties of peptide structures will permit us to develop chemicals that are protease-resistant, that readily cross the plasma membrane, and have desirable pharmacokinetic properties (Eichler et al., 1995). A number of examples now exist in which simple peptides have been converted into peptidomimetics (Kieber-Emmons et al., 1997; Adessi et al., 2002; Patch and Barron, 2002).
There also some advantages of peptides over small molecules since peptide interactions with protein targets can be more specific as compared with small molecules. Large-scale synthesis of peptides has been dramatically improved (Bray, 2003). Not only endogenous peptides and their analogs but also exogenous peptides and their analogs are on the market. These contribute to the understanding of the behavior of peptides in serum. Over the last several years, applications of phage-displayed random peptide library technologies to cancer research led to many important discoveries. Improvements in library construction and availability of a variety of phage display formats will further increase the reliability of the techniques. In the near future, peptides originally isolated from phage-displayed random peptide libraries could be used as therapeutics and diagnostics for cancer. We envision that this field will draw more attention in the next few years since the limitation of antibody-based targeting approach has been realized.
Notes
*http://www.affymax.com/pipeline_hematide.html
*http://www.novagen.com
Abbreviations
- mAb:
-
monoclonal antibody
- HIV:
-
human immunodeficiency virus
- pIII:
-
gene III protein
- pVIII:
-
gene VIII protein
- CK2:
-
casein kinase 2
- IL-6:
-
interleukin-6
- IL-6R:
-
interleukin-6 receptor
- VCAM-1:
-
vascular adhesion molecule-1
- VNP:
-
VHSPNKK-modified magnetofluorescent nanoparticle
- IL-11R:
-
interleukin-11 receptor
- MMP:
-
matrix metalloproteinases
References
Adams G. P., Weiner L. M., (2005). Nat. Biotechnol. 23: 1147–1157
Adams G. P., Schier R., McCall A. M., et al. (2001). Cancer Res. 61: 4750–4755
Adermann K., John H., Standker L., Forssmann W. G., (2004). Curr. Opin. Biotechnol. 15: 599–606
Adessi C., Soto C., (2002). Curr. Med. Chem. 9: 963–78
Aina O. H., Marik J., Liu R., Lau D. H., Lam K. S., (2005). Mol. Cancer Ther. 4: 806–813
Aina O. H., Sroka T. C., Chen M. L., Lam K. S., (2002). Biopolymers 66: 184–199
Akerman M. E., Chan W. C., Laakkonen P., Bhatia S. N., Ruoslahti E., (2002). Proc. Natl. Acad. Sci. U.S.A. 99: 12,617–12,621
Arap W., Haedicke W., Bernasconi M., et al. (2002a). Proc. Natl. Acad. Sci. U.S.A. 99: 1527–1531
Arap W., Kolonin M. G., Trepel M., et al. (2002b). Nat. Med. 8: 121–127
Arap W., Pasqualini R., Ruoslahti E., (1998). Science 279: 377–380
Azzazy H. M., Highsmith W. E. Jr., (2002). Clin. Biochem. 35: 425–445
Barbas III C. F., Burton D. R., Scott J. K., Silverman G. J., (2001). Phage Display: A Laboratory Manual Cold Spring Harbor Laboratory Press Cold Spring Harbor, New York
Bray B. L., (2003). Nat. Rev. Drug Discov. 2: 587–593
Castagnoli L., Zucconi A., Quondam M., et al. (2001). Comb. Chem. High Throughput Screen 4: 121–133
Clackson T., Lowman H. B., (2004). Phage Display – A Practical Approach Oxford University Press New York
Cortez-Retamozo V., Backmann N., Senter P. D., et al. (2004). Cancer Res. 64: 2853–2857
Deshayes K., Schaffer M. L., Skelton N. J., et al. (2002). Chem. Biol. 9: 495–505
Eckert D. M., Malashkevich V. N., Hong L. H., Carr P. A., Kim P. S., (1999). Cell 99: 103–115
Eichler J., Appel J. R., Blondelle S. E., et al. (1995). Med. Res. Rev. 15: 481–496
Ellerby H. M., Arap W., Ellerby L. M., et al. (1999) Nat. Med. 5: 1032–1038
Essler M., Ruoslahti E., (2002). Proc. Natl. Acad. Sci. U.S.A. 99: 2252–2257
Ferrari M., (2005). Nat. Rev. Cancer. 5: 161–171
Fleming T. J., Sachdeva M., Delic M., et al. (2005) J. Mol. Recognit. 18: 94–102
Grifman M., Trepel M., Speece P., et al. (2001) Mol. Ther. 3: 964–975
Haubner R., Wester H. J., (2004). Curr. Pharm. Des. 10: 1439–1455
Ho I. A., Lam P. Y., Hui K. M., (2004). Hum. Gene Ther. 15: 719–732
Hoffman J. A., Giraudo E., Singh M., et al. (2003). Cancer Cell. 5: 383–391
Holig P., Bach M., Volkel T., et al. (2004). Protein Eng. Des. Sel. 17: 433–441
Huang C., Liu X. Y., Rehemtulla A., Lawrence T. S., (2005). Int. J. Radiat. Oncol. Biol. Phys. 62: 1497–1503
Jiang T., Olson E. S., Nguyen Q. T., et al. (2004). Proc. Natl. Acad. Sci. U.S.A. 101: 17,867–17,872
Joyce J. A., Laakkonen P., Bernasconi M., Bergers G., Ruoslahti E., Hanahan D., (2003) Cancer Cell 4: 393–403
Kay B. K., Kurakin A. V., Hyde-DeRuyscher R., (1998) Drug Discov. Today 3: 370–378
Kay B. K., Kasanov J., Yamabhai M., (2001). Methods 24: 240–246
Kayushin A. L., Korosteleva M. D., Miroshnikov A. I., et al. (1996). Nucleic Acids Res. 24: 3748–3755
Kelly K. A., Allport J. R., Tsourkas A., et al. (2005). Circ. Res. 96: 327–336
Kemp, P., Garcia, L. R. and Molineux, I. J.: 2005, Virology 340: 307–317
Kieber-Emmons T., Murali R., Greene M. I., (1997). Curr. Opin. Biotechnol. 8: 435–441
Kolonin M., Pasqualini R., Arap W., (2001). Curr. Opin. Chem. Biol. 5: 308–313
Krag D. N., Fuller S. P., Oligino L., et al. (2002). Cancer Chemother. Pharmacol. 50: 325–332
Kuhnast B., Bodenstein C., Haubner R., et al. (2004). Nucl. Med. Biol. 31: 337–344
Laakkonen P., Akerman M. E., Biliran H., et al. (2004) Proc. Natl. Acad. Sci. U.S.A. 101: 9381–9386
Laakkonen P., Porkka K., Hoffman J. A., Ruoslahti E., (2002) Nat. Med. 8: 751–755
Ladner R. C., Sato A. K., Gorzelany J., de Souza M., (2004). Drug Discov. Today 9: 525–529
Lam K. S., Salmon S. E., Hersh E. M., Hruby V. J., Kazmierski W. M., Knapp R. J., (1991) Nature 354: 82–84
Landon L. A., Deutscher S. L., (2003). J. Cell Biochem. 90: 509–517
Levene A. P., Singh G., Palmieri C., (2005). J. R. Soc. Med. 98: 146–152
Lin M. Z., Teitell M. A., Schiller G. J., (2005). Clin. Cancer Res. 11: 129–138
Liu L., Anderson W. F., Beart R. W., Gordon E. M., Hall F. L., (2000) J. Virol. 74: 5320–5328
Liu, Y.: 2005, Cancer Lett. In press, Corrected Proof.
Matthews T., Salgo M., Greenberg M., et al. (2004). Nat. Rev. Drug Discov. 3: 215–225
Miljanich G. P., (2004). Curr. Med. Chem. 11: 3029–3040
Mori T., (2004). Curr. Pharm. Des. 10: 2335–2343
Nielsen L. L., Young A. A., Parkes D. G., (2004). Regul. Pept. 117: 77
Nilsson F., Tarli L., Viti F., Neri D., (2000). Adv. Drug Deliv. Rev. 43: 165–196
Nixon A. E., (2002). Curr. Pharm. Biotechnol. 3: 1–12
Oku N., Asai T., Watanabe K., et al. (2002). Oncogene 21: 2662–2669
Pasqualini R., Koivunen E., Kain R., et al. (2000). Cancer Res. 60: 722–727
Pasqualini R., Ruoslahti E., (1996). Nature 380: 364–366
Patch J. A., Barron A. E., (2002). Curr. Opin. Chem. Biol. 6: 872–877
Perea S. E., Reyes O., Puchades Y., et al. (2004). Cancer Res. 64: 7127–7129
Petrenko V. A., Smith G. P., (2000). Protein Eng. 13: 589–592
Rafii S., Avecilla S. T., Jin D. K., (2003). Cancer Cell. 4: 331–333
Reilly R. M., Sandhu J., Alvarez-Diez T. M., et al. (1995). Clin. Pharmacokinet. 28: 126–142
Ritchie B. C., (2003). Transfus Apher. Sci. 29: 259–267
Rodi D. J., Makowski L., (1999). Curr. Opin. Biotechnol. 10: 87–93
Rodi D. J., Mandava S., Makowski L., (2004). Bioinformatics 20: 3481–3489
Rodi D. J., Soares A. S., Makowski L., (2002). J. Mol. Biol. 322: 1039–1052
Roges O. A., Baron M., Philis-Tsimikas A., (2005). Expert. Opin. Investig. Drugs 14: 705–727
Romanov V. I., (2003). Curr. Cancer Drug Targets 3: 119–129
Rosenberg A., Griffin K., Studier W., et al. (1996). InNovations 6: 1–6
Ruoslahti E., (2004). Biochem. Soc. Trans. 32: 397–402
Russel, M., Lowman, H. B. and Clackson, T.: 2004, in T. Clackson and H. B. Lowman (eds.), Phage Display-A Practical Approach, Oxford University Press, New York, pp. 1–26
Schumacher T. N., Mayr L. M., Minor D. L. Jr., Milhollen M. A., Burgess M. W., Kim P. S., (1996) Science 271: 1854–1857
Scott, J. K.: 2001, in Barbas III, C. F. et al. (eds.), Phage Display: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 4.1–4.13
Shadidi M., Sioud M., (2004). Methods Mol. Biol. 252: 569–580
Smith G. P., (1985). Science 228: 1315–1317
Stern M., Herrmann R., (2005). Crit. Rev. Oncol. Hematol. 54: 11–29
Su J.-L., Lai K.-P., Chen C.-A., et al. (2005). Cancer Res. 65: 4827–4835
Sullivan D. C., Ferrari M., (2004). Mol. Imaging 2004(3): 364–369
Szardenings M., (2003). J. Recept. Signal Transduct. Res. 23: 307–349
Tian F., Tsao M. L., Schultz P. G., (2004). J. Am. Chem. Soc. 126: 15,962–15,963
Wark P. A., (2002). IDrugs 5: 586–589
Wiesehan K., Buder K., Linke R. P., et al. (2003). Chembiochem. 4: 748–753
Wiesehan K., Willbold D., (2003). Chembiochem. 4: 811–815
Winquist R. J., Pan J. Q., Gribkoff V. K., (2005). Biochem. Pharmacol. 70: 489–499
Xie J., Schultz P. G., (2005) Methods 36: 227–238
Yao V. J., Ozawa M. G., Trepel M., Arap W., McDonald D. M., Pasqualini R., (2005). Am. J. Pathol. 166: 625–636
Zitzmann S., Mier W., Schad A., et al. (2005). Clin. Cancer Res. 11: 139–146
Zurita A. J., Arap W., Pasqualini R., (2003). J. Control Release 91: 183–186
Zurita A. J., Troncoso P., Cardo-Vila M., et al. (2004). Cancer Res. 64: 435–439
Acknowledgements
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors wish to thank Martha Welch, in the department of Scientific Publications, Graphics & Media, Science Applications International Corporation-Frederick, Inc., for assistance with diagrams.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Krumpe, L.R., Mori, T. The Use of Phage-Displayed Peptide Libraries to Develop Tumor-Targeting Drugs. Int J Pept Res Ther 12, 79–91 (2006). https://doi.org/10.1007/s10989-005-9002-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-005-9002-3