
Abstract
Myosin VI (MVI) is the only known myosin walking towards minus end of actin filaments and is believed to play distinct role(s) than other myosins. We addressed a role of this unique motor in secretory PC12 cells, derived from rat adrenal medulla pheochromocytoma using cell lines with reduced MVI synthesis (produced by means of siRNA). Decrease of MVI expression caused severe changes in cell size and morphology, and profound defects in actin cytoskeleton organization and Golgi structure. Also, significant inhibition of cell migration as well as cell proliferation was observed. Flow cytometric analysis revealed that MVI-deficient cells were arrested in G0/G1 phase of the cell cycle but did not undergo increased senescence as compared with control cells. Also, neither polyploidy nor aneuploidy were detected. Surprisingly, no significant effect on noradrenaline secretion was observed. These data indicate that in PC12 cells MVI is involved in cell migration and proliferation but is not crucial for stimulation-dependent catecholamine release.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Highly specialized chromaffin cells of adrenal medulla contain secretory organelles, termed as chromaffin granules, containing catecholamines, nucleotides and several neuropeptides, which are released upon stimulation of nicotinic receptors. The granules (diameter of about 200–300 nm) are formed at the Golgi apparatus and are transported to the cytoplasm where they are stored in two major pools, the reserve and release-ready ones (Hook and Metz-Boutigue 2002; Trifaro 2002). Stimulation leads to translocation of the release-ready pool to the submembrane area, fusion of granules with the plasma membrane, and subsequent release of the granule content. The granule pools are separated by a dense actin filament barrier that, in response to cell stimulation, undergoes precisely regulated depolymerization thus enabling shift of the granules from the reserve to the release-ready pool, and the secretion process (Bader et al. 2002). These and other observations (Giner et al. 2005; Jahn et al. 2003; Lang et al. 2000; Rose et al. 2003; Rudolf et al. 2003; Sasakawa et al. 2002) confirm engagement of actin-based cytoskeleton and myosins in granules transport and secretion of granule content.
Several myosin isoforms including myosins IA, IIA, IIB, VA and VI were detected in chromaffin cells (Wagner et al. 1992; Rose et al. 2002; Wu et al. 2002; Rudolf et al. 2003; Neco et al. 2004; Majewski et al. 2010). Rose et al. (2003) and Rudolf et al. (2003) demonstrated that myosin VA was involved in granule translocation towards the plasma membrane, while lack of functional protein led to a decrease of noradrenaline release and strong reduction of granule motility within the actin cortex. Myosin IIB was not found in association with chromaffin granules, however, its indirect implication in the transport of vesicles and the activity of the docked granules has been postulated (Rose et al. 2003). We showed that myosin VI (MVI) was ubiquitously expressed in PC12 cells and was associated with various vesicular structures and organelles, including chromaffin granules and the nucleus (Majewski et al. 2010).
MVI is one of the unconventional myosins. It consists of a 140-kDa heavy chain containing one IQ motif, to which calmodulin binds. Its C-terminal tail forms a globular domain, essential for cargo binding and/or interaction with binding partners (Sweeney and Houdusse 2007; Altman et al. 2007; Buss and Kendrick-Jones 2011). The recombinant full-length protein is a monomer but it is not excluded that in vivo MVI may function as a dimer (Lister et al. 2004; Noguchi et al. 2009). Numerous observations suggest that MVI is involved in vesicular transport (Mermall et al. 1994; Buss et al. 2002). It is postulated that the C-terminal globular domain binds to membrane-associated proteins (termed as MVI binding partners), such as Dab2 in small intestine epithelial cells (Morris et al. 2002), SAP97 in the central nervous system neurons (Wu et al. 2002), GIPC in retinal pigment epithelium (Aschenbrenner et al. 2003) or optineurin (Spudich et al. 2007). Also, it was shown that the positively charged region of the globular tail binds specifically to PIP2-containing liposomes (Spudich et al. 2007).
MVI seems to be a processive motor walking on actin filaments; however, unlike other myosins characterized so far, it moves towards the minus (pointed) end of actin filaments (Wells et al. 1999). It implies that it may play different role than other myosins engaged in the same processes, for example in intracellular trafficking of organelles and protein complexes. Mutations within the MVI gene lead to deafness in mice (Snell’s waltzer syndrome) and humans due to disintegration of the inner ear hair cells (see Redowicz 2002). There are also other symptoms in mice such as head tossing and hyperactivity, and in humans gradually developing blindness, craniofacial aberrations and hypertrophic cardiomyopathy. Further studies have revealed that Golgi morphology was affected and collagen secretion was reduced in fibroblasts derived from the Snell’s waltzer mouse (Warner et al. 2003). Moreover, defects in CFTR (cystic fibrosis transmembrane conductance regulator) apical endocytosis and morphology of brush border enterocytes as well as renal proximal tubular endocytosis were observed (Ameen and Apodaca 2007; Gotoh et al. 2010). Also, severe defects were visible in hippocampus and the hippocampal neurons displayed a significant deficit in the stimulation-induced internalization of glutamate receptors (Osterweil et al. 2005). Moreover, Yano et al. (2006) have demonstrated that MVI in hippocampal neurons is involved in neutrotransmission dependent on brain-derived neurotrophic factor (BDNF) and its receptor (TrkB).
Functional studies and cellular localization indicate that MVI plays a major role in endocytic traffic pathways as well as in cell motility (see Sweeney and Houdusse 2007; Buss and Kendrick-Jones 2008). MVI is localized to clathrin coated pits/vesicles (Buss et al. 2001), the Golgi complex (Warner et al. 2003), membrane ruffles at the leading edge (Buss et al. 1998) and pre- and postsynaptic structures in neurons (Osterweil et al. 2005). It has been also demonstrated that MVI is directly involved in the AP-1B-dependent sorting of proteins to the basolateral plasma membrane in polarized epithelial cells (Au et al. 2007). The exact role of MVI in endocytosis still needs to be established as it is not clear whether it works as the transporting motor or as the anchor linking vesicles and/or plasma membrane proteins to the actin cytoskeleton, responsible for maintenance of cytoskeleton compartments integrity. In addition to its role in membrane trafficking pathways, results obtained on Drosophila MVI in the ovary border cells and spermatids (Hicks et al. 1999; Geisbrecht and Montell 2002) and its localization to membrane ruffles in mammalian fibroblasts (Buss et al. 1998) indicate that MVI is also essential for cell locomotion. Furthermore, observations that MVI is overexpressed in the malignant human ovarian cancer cells and that inhibition of its expression in the mouse tumor cells with the RNAi significantly inhibits their invasiveness (Yoshida et al. 2004) may also confirm its role in cell migration.
In this study, we addressed a role of MVI in PC12 cells derived from rat pheochromocytoma (adrenal medulla tumor), which are widely used as a cell model to study neuronal and endocrine secretion (Martin and Grishanin 2003). The data indicate that MVI is involved in cell migration and proliferation but is not crucial for catecholamine secretion.
Materials and methods
Cell culture
PC12 cells (American Cell Culture Collection, USA) were cultured at 37°C under 5% CO2 in RPMI 1640 media supplemented with 10% heat-inactivated horse serum and 5% fetal bovine serum or F12K media supplemented with 15% heat-inactivated horse serum and 2.5% fetal bovine serum.
siRNA knockdown of MVI
PC12 cell lines, in which MVI gene expression was knocked down were created using constructs based on pSilencer 2.1-U6 hygro vector (Ambion Inc., USA) with introduced the following siRNA sequences: (i) for a transient knockdown (T-MVI) 5′-ATCTAAGCTGGCAGTCCAC-3′ (corresponding to 1882–1900 nt; Fig. 1a) and (ii) for a stable knockdown (S-MVI) 5′-GAGAGUCAAUUUGUGGCUA-3′ (corresponding to 2346–2364 nt, Fig. 1a). siRNA sequences against a coding region of rat MVI mRNA were designed using siRNA Target Finder available on line (http://www.ambion.com/techlib/misc/siRNA_finder.html). As negative controls, mock transfected cells or cells transfected with pSilencer vector carrying scrambled shRNA (sc1MVI or sc2MVI) were used. Scrambled shRNA were designed based on the original siRNA sequences using on-line GeneScript tool. The following scrambled sequences were selected: for sc1MVI 5′-GCATTGTGGATGAATAGCT-3′ and for sc2MVI 5′-CGCATTGTGGATGAATAGC-3′. PC12 cells were transfected using Lipofectamine 2000 (Invitrogen, USA). Stably transfected cells (from two independent transfections: SA-MVI and SB-MVI) with reduced levels of MVI expression were selected based on their resistance to hygromycin. Both T-MVI and S-MVI cells were examined for MVI gene expression by the Western blot, and S-MVI cell lines by RT-PCR technique. To amplify fragments of MVI as well as of GADPH genes (used as the internal control to normalize gene expression), the following primers were used: MVI_F 5′-GATGGAGCTGCACCCTGACA-3′, MVI_R 3′-GCTCTCAATGGCGCTCTGAAG-5′ and GAPDH_F 5′-ACCACAGTCCATGCCATCAC-3′, GAPDH_R 3′-TCCACCACCCTGTTGCTGTA-5′.

Depletion of MVI by siRNA technique affects morphology of PC12 cells. a Diagram presenting the location of the regions to which shRNA oligos were produced to obtain cell lines with transient MVI knockdown (T-MVI cells) and stable MVI knockdown (S-MVI cells); b Immunoblot analysis of MVI level in lysates of cells transiently transfected with pSilencer vector to knockdown MVI gene expression (T-MVI) as well as lysates of control cells without the vector (mock control) and the vector containing scrambled shRNA sequence (sc1MVI), 24, 48, 72 and 96 h after transfection. Lower panel, a 42-kDa band corresponding to actin was stained with Ponceau red on a nitrocellulose membrane after transfer of the gel with the above described samples. The quantification of MVI content in T-MVI samples in every time-point was made in reference to control sc1MVI lysates at the corresponding time-point. c Immunoblot and RT-PCR analyses of SA-MVI and SB-MVI cells as well as of mock-control cells and sc2MVI cells. Lower panels, a 42-kDa band detected with anti-β-actin antibody and an amplified GAPDH fragment, used as internal controls. The quantification of MVI content in SA-MVI and SB-MVI samples was made in reference to the control sc2MVI sample. For b and c, this is as representation of results obtained from three independent experiments. d Analysis of size of SA-MVI, mock control and sc2MVI cells. Cells were stained with TRITC-phalloidin and DAPI, and then cell area was estimated with Leica software; bar 50 μm. The results are presented for 150 cells from each cell type, where 100% represents the sum of the cells of a given line subjected to the analysis. Statistical relevance measured with Student’s t test *P < 0.05 and ***P < 0.001, respectively. e Scanning electron microscopy images of examples of SA-MVI as well as mock control and sc2MVI cells. Bars 5 μm. All other details as described under “Materials and methods” section
Antibodies and fluorescent markers
Rabbit polyclonal antibody directed against amino-acid residues 1049–1054 of porcine MVI heavy chain, originally developed by Dr. Tama Hasson, was from Proteus (USA). Rabbit polyclonal antibody against myosin V (MV) and monoclonal antibody against β-actin were purchased from Sigma-Aldrich (USA). Monoclonal antibodies against GM130 and TGN38 as well as against dopamine β-hydroxylase (DBH) were from BD Transduction Laboratories (USA). DAPI (4α, 6-diamidino-2-phenylindole) was from Vector Laboratories (USA). TRITC-phalloidin as well as To-Pro3 were from Invitrogen (USA). For immunocytochemistry studies, the following secondary antibodies from Molecular Probes were used: goat anti-rabbit IgG labeled with Alexa Fluor 488 dye and goat anti-mouse IgG labeled with Alexa Fluor 546 dye.
Stimulation of secretion
To induce noradrenaline (NA) secretion, PC12 cells were cultured as described above and stimulated essentially according to Trifaró and Lee (1980) and Vitale et al. (1992). Briefly, cells were washed with Locke’s solution containing 2.6 mM KCl, 154 mM NaCl, 2.2 mM CaCl2, 0.5 mM KH2PO4, 1.25 mM K2HPO4, 1.2 mM MgCl2 and 10 mM glucose. Then, they were incubated for 5–10 min in Locke’s solution with elevated K+ concentration (56 mM KCl, 103.6 mM NaCl, 2.2 mM CaCl2, 0.5 mM KH2PO4, 1.25 mM K2HPO4, 1.2 mM MgCl2, 10 mM glucose) to stimulate the secretion, or in calcium-free Locke’s solution (154 mM NaCl, 2.6 mM KCl, 0.5 mM KH2PO4, 1.25 mM K2HPO4, 1.2 mM MgCl2 and 10 mM glucose) to block the secretion. The cells were further processed for immunocytochemistry, electron microscopy or catecholamine release assay.
Cell lysates
Cells were washed with ice-cold PBS, collected at 800 x g and homogenized in a Teflon homogenizer in a buffer [50 mM Tris–HCl pH 8.0; 150 mM NaCl, 0.1% TritonX-100, 2 mM EGTA, 1 mM DTT, 1 mM PMSF and Complete protease inhibitor cocktail (Roche Diagnostics GmbH, Germany)]. Lysates were subjected to the SDS-PAGE followed by the immunoblot analysis of the presence of MVI using anti-MVI and anti-βactin antibodies. Protein concentration was determined using the standard Bradford method.
Immunoblotting
PC12 cell lysates were separated using 10% polyacrylamide SDS gels and then transferred to a nitrocellulose membrane. After transfer, the membrane was blocked for 1 h at room temperature in TBS containing 5% non-fat milk powder and 0.2% Triton X-100 followed by 1-h incubation with appropriate dilutions of different antibodies described above. The primary antibodies were detected using a 1:10000 dilutions of anti-rabbit or 1:5000 anti-mouse secondary antibodies conjugated with horse radish peroxidase; the reaction was developed using the ECL method as described by the manufacturer (Pierce, USA). Usually 10–20 μg of protein was loaded and 42-kDa band corresponding to actin stained either with Coomassie brilliant blue in the gels or with Ponceau red on the nitrocellulose, or detetected with anti-β-actin antibody served as the internal loading control.
Densitometric analysis
The content of MVI or RT-PCR product corresponding to MVI cDNA fragment was estimated by densitometric analysis of immunoblots and agarose gels, respectively. The membranes or gels were photographed using G:Box system from SynGene (Cambridge, Great Britain) equipped with GeneSnap and GeneTools software.
Immunolocalization studies
Distribution of MVI and other marker proteins in PC12 cells was examined by indirect immunocytochemistry before or after stimulation with 56 mM KCl. Cells were fixed in 4% paraformaldehyde for 20 min and then permeabilized with 0.1% Triton X-100 for 5 min. The fixed specimens were thoroughly washed in phosphate-buffered saline (PBS) and treated for 1 h with 3% solution of bovine serum albumin (BSA) in PBS. Cells were incubated overnight at 4°C with anti MVI antibodies at a dilution of 1:50. This was followed by incubation with Alexa 488-conjugated secondary anti-rabbit antibodies at a dilution of 1:1000 for 1 h. For simultaneous assessment of distribution of other proteins, cells were also incubated with appropriate dilutions of the above described antibodies, and then with Alexa 546-conjugated secondary antibodies. The specimens were visualized using Leica TCS SP2 or Leica TCS SP5 spectral confocal microscopes.
Scanning electron microscopy (SEM)
For SEM, cells were fixed in 2.5% glutaraldehyde with 0.1% tannic acid in PBS for 1 h, dehydrated through graded series of ethanol and acetone, dried by CO2 critical point method and then coated with a thin layer of carbon and gold. Samples were examined using Jeol JEM-1200EX electron microscope with an ASID 19 scanning attachment, operating at 80 kV.
Transmission electron microscopy (TEM)
Cells were fixed in 0.5% glutaraldehyde with 2.5% paraformaldehyde in 0.2 M phosphate buffer pH 7.2 for 1 h, dehydrated through graded series of ethanol and embedded in LR White resin from Fluka (Switzerland). The samples were polymerized at 55°C for 48 h and cut on ultramicrotome. Ultrathin sections were placed on copper grids and analyzed under a JEOL JEM-1200EX electron microscope.
Evaluating cell size and proliferation
After 48 h S-MVI, sc2MVI and mock control PC12 cells were fixed in 4% paraformaldehyde for 20 min and washed twice with PBS. Next, they were quenched for 30 min in PBS with 50 mM NH4Cl and incubated for 1 h in blocking solution (2% horse serum in PBS/0.02% Triton X-100). To visualize actin cytoskeleton and nuclei, the cells were stained for 15 min with 50 mg/ml of TRITC-conjugated phalloidin (Sigma, USA) and To-Pro3 (Invitrogen, USA) in PBS, and next washed three times with PBS before mounting. Immunofluorescent images were prepared using laser scanning confocal microscope Leica TCS SP2 (Leica Microsystems). Next, at least 150 single cells for each the cell type were outlined using the ImageJ software and their area was calculated, using the same software. Statistical analysis was performed using Student’s t test.
For the proliferation assay, cells were grown up to 96 h and counted either in the Neubauer chamber or using hand-held automated cell counter (Scepter 2.0, Millipore, USA). They were plated onto six-well dishes at the density of 1 × 105 cells per well. Next after 72 and 96 h, cells were collected by trypsinization, counted and the population doubling time (DT) was calculated. To estimate cell viability, either trypan blue standard technique or flow cytometry cell viability assay using propidium iodide were employed. Statistical analysis was performed using Student’s t test.
Catecholamine release
Secretion assays were performed according to Francis et al. (2002) with modifications. PC12 cells with MVI knockdown (T-MVI) and control sc1MVI were cultured for 72 h on 24-well plates in culture medium with 100 nM [3H]noradrenaline ([3H]NA; 54.6 Ci/mmol, Perkin Elmer, Boston, USA) and 100 nM nonradioactive noradrenaline (NA) for 10 min at 37°C, exhaustively washed with Locke’s solution and then incubated for 7 min at 37°C in Locke’s solution with elevated K+ concentration to stimulate the secretion, or in calcium-free Locke’s solution to block the secretion. Next, the cells were washed again with Locke’s solution and lysed in 5% SDS. All samples: media, solutions as well as cell lysates were collected and the amount of [3H]NA in the samples was determined by scintillation counter (Beckman LS 6500, USA) to calculate the NA release and uptake. The ratios of NA release to uptake in control and transfected cells were quantified in four independent experiments performed in duplicates.
Wound healing assay
A scratch was made with a 0.2 ml pipette tip across a monolayer of SA-MVI and SB-MVI cells as well as sc2MVI and mock control PC12 cells, which were cultured on 60-mm dish. Cell debris was washed off and a new portion of culture medium was added. Images of five different areas along the artificial wound were made after 24 and 48 h culture, and the number of cells detected in the scratch area was counted. Statistical analysis was performed using Student’s t test.
Migration assay
To assess the effect of MVI knockdown on migration of PC12 cells, 5000 of SA-MVI cells as well as control cells: sc2MVI and mock control were plated on 8-well chambered coverglass (Nunc) and cultured for 24 h. Cells were filmed in bright field for 12 h with 10 min time lapse using Leica TCS SP5 confocal microscope with 10× objective and a temperature-controlled live-cell imaging chamber. At least 20 cells from each cell type were tracked using MetaMorph software in each of three independent experiments. Statistical analysis was performed using Student’s t test.
Detection of senescence-associated β-galactosidase
SA-β-galactosidase (SA-β-gal) activity was measured using flow cytometry, as described by Kurz et al. (2000). Briefly, SA-MVI PC12 cells and the control ones (sc2MVI and mock control) were incubated with 33 mM non-fluorescent SA-β-gal substrate (C12FDG; Invitrogen, Paisley, UK) and after exhaustive wash the fluorescence corresponding to the product SA-β-galactosidase was immediately measured by flow cytometry using CellQuest Software; 10 000 events were counted for each sample (FACS Calibur, Becton–Dickinson).
DNA content analysis and cell cycle analysis
For DNA content analysis, the culture medium was discarded and SA-MVI and control PC12 cells were collected by trypsinization, fixed in 70% ethanol overnight at 4°C and then stained with PI solution (3.8 mM sodium citrate, 50 mg/ml RNAse A and 500 μg/ml propidium iodide in PBS). DNA content was determined by flow cytometry using CellQuest Software; 10.000 events were counted for each sample (FACSCalibur, Becton–Dickinson). The percentage of cells in a particular cell cycle stage was calculated by the ModFit software (Becton–Dickinson, USA).
Results
Analysis of PC12 cell lines with MVI expression knockdown
We have previously shown that in PC12 cells MVI is ubiquitously expressed and is associated with various vesicular structures, including synaptic vesicles and chromaffin granules (Majewski et al. 2010).
To characterize a role of MVI in PC12 cells, siRNA technique was employed to knockdown its expression and to produce cell lines: one with transiently (T-MVI) and two independent with stably (SA-MVI and SB-MVI) knocked down MVI expression (Fig. 1a). Figure 1b demonstrates Western blot analysis of T-MVI cells lysates, in comparison with mock transfected cells (control), and cells transfected with scrambled MVI sequence (sc1MVI). MVI level in T-MVI cells was decreased by about 70–80% 96 h after the transfection in comparison with control cells. Similar inhibition was achieved for SA-MVI and SB-MVI cell lines in regard to sc2MVI control cells (Fig. 1c). Protein level was decreased by about 80% for SA-MVI and 95% for SB-MVI, and RNA level was decreased by about 60% and 80%, respectively. Both stably transfected cell lines exhibited nearly identical properties therefore in most of the cases only one of them is presented herein.
Analysis of SA-MVI cell line by means of the fluorescence and scanning electron microscopy revealed the presence of significant pools of small and large cells, contrary to mock control cells and sc2MVI cells (Fig. 1d, e). Quantification of the cell area revealed that for mock control cells it was 825 ± 403 μm2, for sc2MVI cells—1009 ± 550 μm2 while for SA-MVI cells—1416 ± 2078 μm2. As presented in Fig. 1d, there was about twice as much of large (size >1500 μm2) and small cells (size <400 μm2) in SA-MVI cell line in comparison with mock and sc2MVI control cells. The morphology of SA-MVI cells, especially of those large ones, was also changed as presented in Fig. 1e. The cells became more flat, less polarized, and seemed to form multiple thin filopodia-like processes.
Effect of myosin VI depletion on organization of Golgi apparatus and actin cytoskeleton
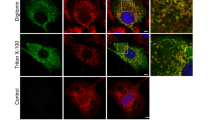
Warner et al. (2003) showed for the first time that in fibroblasts derived from Snell’s waltzer mice lacking MVI, profound changes in Golgi organization were observed. We also observed severe alterations in Golgi morphology in both T-MVI (Fig. 2a) and both S-MVI cell lines (Fig. 2b, presenting SA-MVI cells). To visualize the Golgi complexes in SA-MVI cells, antibodies against two Golgi protein markers were used, GM130 and TGN38. While in both types of control cells Golgi apparatus was concentrated in well organized compact structures adjacent to a small fraction of the nucleus circumference, in MVI-deficient, T-MVI and SA-MVI cells Golgi complexes were fragmented and elongated, often forming a semi-coat around the nucleus (Fig. 2a, arrowheads and b). These data suggest that also in PC12 cells MVI is engaged in Golgi organization.

Depletion of MVI affects Golgi and cytoskeleton organization. MVI distribution in T-MVI 72 h after transfection (a) and SA-MVI cells (b) as well as in control cells (mock control and sc1MVI or sc2MVI) was visualized with anti-porcine MVI antibody (green). Golgi structures were stained with anti-GM130 or TGN38 antibodies (in red), microfilaments with TRITC-phalloidin (red) and nuclei with ToPro3 (blue). In a, arrowheads point to the most altered Golgi complexes. These are the 0.8-μm images of the cell center attained with Leica TCS SP2 or SP5 spectral confocal microscopes equipped with HCX PL APO 63x/1.25-0.75 Oil Cs objectives. Other details as described under “Materials and methods” section. Bars 10 μm
MVI is believed to be involved in the regulation of actin cytoskeleton organization (Hertzano et al. 2008; Chibalina et al. 2009, 2010; Jbireal et al. 2010; Mangold et al. 2011). We tested whether MVI depletion affected organization of actin cytoskeleton; as e.g. in a large S-MVI cell shown in Fig. 2b. In control cells, filamentous actin localized in the cell cortex and few stress fibers were visible. In MVI depleted cells (here shown SA-MVI cells), actin was seen as the thick cortical layer and in numerous filopodial structures. It was also present in the perinuclear area and big spots around large vacuolar structures. These data suggest that MVI is engaged in cytoskeleton organization also in PC12 cells.
Effect of MVI depletion on PC12 cells migration
Changes in actin cytoskeleton are known to affect cell motility. Therefore we decided to test whether depletion of MVI affected S-MVI cells migration (Fig. 3a–c).

Depletion of MVI affects migration and proliferation of PC12 cells. a Wound healing assay of SA-MVI and SB-MVI cells as well as mock control and sc2MVI cells. Images of the same scratch areas were taken after 24 and 48 h, as indicated. The dashed line represents the initial scratch and measurement area. These are representative images from five independent experiments. b Quantification of cell number from experiments described in “a”. The values are the means (±SD) of the cells counted in the measurement area. Statistical relevance measured with Student’s t test is ***P < 0.001. c Migration rates of SA-MVI, sc2MVI and mock control cells assessed with time lapse technique of living cells and quantified with Metamorph software. At least 20 cells from each line were analyzed in each of three independent experiments. The values are the means (±SD); statistical relevance measured with Student’s t test is *P < 0.05. d Proliferation test of SA-MVI and SB-MVI as well as of control sc2MVI and mock control cells. The number of cells in each of the cell culture was counted after 72 and 96 h. The population doubling time (DT) for each of the culture was calculated based on the data obtained from three independent experiments (n = 3); statistical relevance measured with Student’s t test is ***P < 0.001. e Viability test of SA-MVI and SB-MVI as well as of control sc2MVI and mock transfected cells. Cells were subjected to the flow cytometric analysis using propidium iodide after 48, 72 and 96 h of culture. The values are obtained from two independent experiments; Other details as described under “Materials and methods” section
Using wound healing assay it was shown that the number of cells migrating into the wound area after 24 h was over thrice smaller for SA-MVI and SB-MVI cells in comparison with the control cells (21 ± 2 cells in the scratch zone for SA-MVI and 19 ± 2 for SB-MVI vs. 65 ± 3 cells for mock control and 55 ± 3 for sc2MVI). Similar observation was made after 48 h (59 ± 3 cells for SA-MVI and 45 ± 4 for SB-MVI vs. 158 ± 5 for mock control and 154 ± 6 for sc2MVI).
These results indicate that MVI may be involved in PC12 cells motility, thus confirming observations of Geisbrecht and Montell (2002) and Yoshida et al. (2004), who demonstrated that MVI was engaged in the migration of Drosophila border cells and malignant human ovarian cancer cells, respectively. It is known that wound healing is dependent not only on cell migration but also on cell proliferation, especially for longer incubation times. Therefore, the time lapse technique with tracking of individual cells was applied to assess migration of the living cells (Fig. 3c). Depletion of MVI caused statistically significant changes in migration rate (0.29 ± 0.07 μm/min for SA-MVI cells) in regard to mock control (0.40 ± 16 μm/min) and sc2MVI (0.39 ± 15 μm/min) cells thus confirming involvement of this motor protein in migration of PC12 cells.
To test whether MVI depletion affects also proliferation of PC12 cells, a defined number (1 × 105) of both control and SA-MVI and SB-MVI cells was cultured for 96 h and at the given time intervals aliquots were taken and the cells were counted (Fig. 3d). As seen on the graph, depletion of MVI caused a significant decrease in cell number in both S-MVI cell lines. While in both types of control cells after 96 h it was about 10 × 106 cells, in S-MVI cells it was about 5 × 106. Also, the doubling time (time required to double cell number in a culture) was almost twice longer for the S-MVI cells: for both types of control cells it was 25.4 ± 1.2 h for mock control and 27.2 ± 1.1 h for sc2MVI while it was 44.4 ± 5.2 h for SA-MVI and 45.3 ± 3.6 h for SB-MVI cells. These values were not affected by PC12 cell viability as it was nearly the same for all tested cell types (96–99%) as shown in Fig. 3e.
These data indicate that in PC12 cells MVI functions not only in cell locomotion but may be also engaged in regulation of cell proliferation.
Effect of MVI depletion on PC12 cell cycle
The observed differences in cell proliferation indicate that MVI may be involved in regulation of cell division. To address this, we performed quantification of the DNA content in both types of control cells and S-MVI cells by means of flow cytometry (Fig. 4).

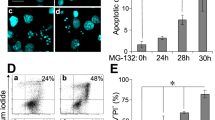
MVI and the PC12 cell cycle. a Flow cytometric analysis of cell cycle of SA-MVI, mock transfected and sc2MVI cells. DNA content was estimated using propidium iodide (left panels) and cell cycle analysis (right panels) was made with Modtif software (Becton–Dickinson). These are representative histograms and analyses from three independent experiments. b SA β-galactosidase activity measurements of SA-MVI and mock transfected and sc2MVI cells. The bars represent the mean values (±SD) of fluorescence corresponding to SA β-galactosidase product. The values were obtained from three independent experiments. Other details as described under “Materials and methods” section
Analysis of the DNA content revealed there were no major differences between S-MVI and control cells, neither aneuploidy nor polyploidy (Fig. 4a). However, we found that MVI depleted cells were arrested in G0/G1 phase of the cell cycle, as the percentage of SA-MVI cells entering G2/M phase was about four times smaller than in control cells (5.75% for SA-MVI vs. 25.22% for mock control and 18.64 for sc2MVI).
Noteworthy, the observed changes in cell proliferation, cell size and cell cycle arrest in G0/G1 phase are also characteristic for the senescent cells. Another feature of senescent cells is an increase of SA-β-galactosidase (SA-β-gal) activity, which can be measured by flow cytometry. To address whether stable depletion of MVI can promote cell senescence, the cytometric SA-β-gal activity test was performed (Fig. 4b). There were no significant differences in the fluorescence corresponding to the SA-β-gal product between both types of control and SA-MVI cells, thus indicating that depletion of MVI was not evoking cell senescence.
MVI depletion does not affect catecholamine secretion
Observation that in PC12 cells depletion of MVI caused changes in Golgi organization and actin cytoskeleton organization encouraged us to test whether the depletion affects catecholamine secretion. The experiment was performed according to the method of Francis et al. (2002) using radioactive noradrenaline (3H[NA]) and quantification of its uptake and release in samples from control sc1MVI and T-MVI cells (Fig. 5A). In T-MVI cells grown for 72 h, upon stimulation with 56 mM KCl about 10% increase in the ratio of 3H[NA] release:uptake was observed, when compared with the control sc1MVI [Fig. 5A(a)]. However, this increase was statistically insignificant and most probably associated with the higher amount of noradrenaline incorporated by the cells with MVI knockdown [Fig. 5A(b)].

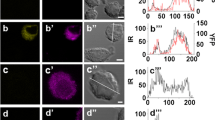
MVI is not crucial for catecholamine secretion. A Influence of MVI knockdown on secretion (a) and uptake (b) of noradrenaline (NA) in T-MVI and control sc1MVI cells. These values (presented as means ± SD) were calculated based on the radioactive noradrenaline ([3H[NA]) measurements in the samples, as described in “Materials and methods”. (a) Secretion stimulated with 56 mM KCl is presented as the ratio of release (amount of radioactivity released to the medium) to uptake, and is referred to non-stimulated both control sc1MVI and knocked down (T-MVI) cells; (b) uptake reflects the amount of radioactivity incorporated into the cells and is a sum of the radioactivity both released to the medium and remained in the cells (control, sc1MVI) and (T-MVI). B Distribution of MVI and MV in PC12 cells before and after 10-min stimulation with 56 mM KCl. Both myosins (in green) were stained with the respective antibodies and chromaffin granules (red) with anti-dopamine β-hydroxylase antibody. These are the 0.8 μm images of cell center attained with Leica TCS SP2 spectral confocal microscope equipped with HCX PL APO 63x/1.25-0.75 Oil Cs objective. Other details as described under “Materials and methods” section. Bars 10 μm. C Morphology of unstimulated and 10-min stimulated PC12 cells. Micrographs were attained with scanning (SEM) and transmission (TEM) electron microscopes as described under “Materials and methods” section. Bars 1 μm in SEM and 200 nm in TEM
Also, we tested whether 10-min stimulation affected MVI localization in control cells (Fig. 5B). There were no substantial changes in its distribution that was in contrast with MV localization, which after 10 min incubation with KCl shifted from the cytoplasm to the cell periphery (Fig. 5B). As expected, upon stimulation the distribution of dopamine β-hydroxylase (the chromaffin granule marker) shifted towards plasma membrane. The stimulation-dependent change in the granule localization was further confirmed by transmission electron microscopy (Fig. 5C, TEM). Also, scanning electron microscopy revealed that stimulation evoked noticeable changes in PC12 cells morphology as the surface of cell body had become more wrinkled (Fig. 5C, SEM).
Discussion
We have shown herein that MVI is involved in cell migration and proliferation but not in catecholamine secretion in neurosecretory PC12 cells. The latter is in contrast with MV, which was shown in numerous studies to play an important role in catecholamine release (Rose et al. 2003; Rudolf et al. 2003; Watanabe et al. 2005).
Our observation that depletion of MVI inhibited PC12 cell migration is in line with the previous studies on Drosophila border cells or malignant human ovarian cancer cells (Geisbrecht and Montell 2002; Yoshida et al. 2004). The authors pointed at cadherin-dependent mechanism as the one, in which MVI could be involved in. Our studies indicate that MVI-dependent regulation of cell migration might also result from its involvement in regulation of actin cytoskeleton organization as significant changes in actin cytoskeleton arrangement in MVI-deficient cells were observed. It is known that several factors such as neurotoxin taipoxin or actin-fragmenting protein scinderin destabilize very dense F-actin rich cortex of PC12 cells thus enhancing neurotransmitter release (Vitale et al. 1991, 1992; Neco et al. 2003; Dumitrescu Pene et al. 2005). Thus it is possible that these MVI-associated cytoskeleton rearrangements might be responsible for the observed inhibition of PC12 cell migration.
We have also observed that depletion of MVI affected PC12 cell proliferation. MVI involvement in the regulation of cell division was reported for the first time by Petritsch et al. (2003) who demonstrated that in Drosophila neuroblasts MVI isoform, Jaguar, was necessary for basal protein targeting and spindle orientation. Arden et al. (2007) have shown that in dividing epithelial MDCK cells localization of MVI changed dramatically and inhibition of its expression by siRNA led to a delay in metaphase progression and a cytokinesis defect. Using flow cytometry techniques, we showed that the observed effect of MVI depletion on PC12 cell proliferation was most probably caused by cell cycle arrest in G0/G1 phase and was not resulting from cell senescence. Yoshida et al. (2004) have reported, however, that inhibition of MVI expression did not affect cell proliferation of malignant ovarian cancer cells. Yet another mechanism of engagement of MVI in the control of cell proliferation was proposed by Vreugde et al. (2006) who demonstrated that MVI was involved in the regulation of gene transcription in human prostate cancer PC3 cells. The presence of MVI in the nuclei of both PC12 and primary culture bovine adrenal medulla cells seems to indicate that this mechanism could be also utilized in these neurosecretory cells (Majewski et al. 2010).
A couple of studies have tied MVI with the secretion process (Warner et al. 2003; Osterweil et al. 2005; Yano et al. 2006). Warner et al. (2003) have shown that fibroblasts obtained from mice lacking MVI did not only have smaller (by ~40% compared with wild-type cells) and altered Golgi complexes but also secreted less of a reporter protein, indicating strong relationship between MVI-dependent functional state of Golgi and secretory properties of fibroblasts. Similar conclusions were reached based on the studies on hippocampal neurons. Yano et al. (2006) showed that MVI played important role in neurotransmission as its lack resulted in 63% decrease of basal synaptic transmission and abolished the BDNF-induced glutamate release. Herein, we also observed significant changes in Golgi complex morphology in transiently and stably MVI depleted cells. Therefore, it was surprising that our studies did not reveal any substantial effect of MVI depletion on noradrenaline release. Instead of expected inhibition of neurotransmitter secretion, we obtained statistically insignificant increase in the release:uptake ratio in the knockdown cells. This small enhancement was most probably due to the increased uptake of radioactive noradrenaline resulting from significant changes in actin cytoskeleton organization observed in MVI-deficient cells. This observation was also inconsistent with studies demonstrating that lack of MVI impaired endocytosis-based incorporation of particles (Osterweil et al. 2005; Ameen and Apodaca 2007; Holt et al. 2007; Gotoh et al. 2010). Noteworthy, we observed that stimulation-dependent translocation of chromaffin granules towards cell periphery was not associated with MVI but with MV, indicating that MVI might not be directly involved in chromaffin granule translocation.
We think that lack of the evident effect of MVI depletion on noradrenaline secretion could be explained by (i) the presence of about 20–30% of endogenous protein in T-MVI cells, which might still compensate for MVI deficit; (ii) changes in actin cytoskeleton organization in T-MVI cell, which enabled higher catecholamine uptake by the modified cells and/or for more efficient transport by the actin-based molecular motors such as MV of chromaffin granules to the cell periphery; (iii) another, yet unrecognized, mechanism in PC12 cells of MVI involvement in catecholamine secretion and endocytosis, and finally (iv) MVI in PC12 cells is not involved in neurosecretion.
Summarizing, our studies addressing the role of MVI in neurosecretory PC12 cells indicate that MVI is involved in actin cytoskeleton organization, cell migration and proliferation as well as in Golgi maintenance but is not a key motor in catecholamine secretion.
References
Altman D, Goswami D et al (2007) Precise positioning of myosin VI on endocytic vesicles in vivo. PLoS Biol 5:e210
Ameen N, Apodaca G (2007) Defective CFTR apical endocytosis and enterocyte brush border in myosin VI-deficient mice. Traffic 8:998–1006
Arden SD, Puri C et al (2007) Myosin VI is required for targeted membrane transport during cytokinesis. Mol Biol Cell 18:4750–4761
Aschenbrenner L, Lee TT et al (2003) Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol Biol Cell 14:2728–2743
Au JSY, Puri C et al (2007) Myosin VI is required for sorting of AP-1B-dependent cargo to the basolateral domain in polarized MDCK cells. J Cell Biol 177:103–114
Bader MF, Holz RW et al (2002) Exocytosis: the chromaffin cell as a model system. Ann NY Acad Sci 971:178–183
Buss F, Kendrick-Jones J (2008) How are the cellular functions of myosin VI regulated within the cell. Biochem Biophys Res Commun 369:165–175
Buss F, Kendrick-Jones J (2011) Multifunctional myosin VI has a multitude of cargoes. Proc Natl Acad Sci USA 108:5927–5928
Buss F, Kendrick-Jones J et al (1998) The localization of myosin VI at the Golgi complex and leading edge of fibroblasts and recruitment into membrane ruffles of A431 cells after growth factor stimulation. J Cell Biol 143:1535–1545
Buss F, Arden SD et al (2001) Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J 20:3676–3684
Buss F, Luzio JP et al (2002) Myosin VI, an actin motor for membrane traffic and cell migration. Traffic 3:851–858
Chibalina MV, Puri C et al (2009) Potential roles of myosin VI in cell motility. Biochem Soc Trans 37:966–970
Chibalina MV, Poliakov A et al (2010) Myosin VI and optineurin are required for polarized EGFR delivery and directed migration. Traffic 11:1290–1303
Dumitrescu Pene T, Rose SD et al (2005) Expression of various scinderin domains in chromaffin cells indicates that this protein acts as a molecular switch in the control of actin filament dynamics and exocytosis. J Neurochem 92:780–789
Francis SC, Sunshine C et al (2002) Coordinate regulation of catecholamine uptake by rab3 and phosphoinositide 3-kinase. J Biol Chem 277:7816–7823
Geisbrecht ER, Montell DJ (2002) Myosin VI is required for E-cadherin-mediated border cell migration. Nat Cell Biol 4:616–620
Giner D, Neco P et al (2005) Real-time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells. J Cell Sci 118:2871–2880
Gotoh N, Yan Q et al (2010) Altered renal proximal tubular endocytosis and histology in mice lacking myosin-VI. Cytoskeleton 67:178–192
Hertzano R, Shalit E et al (2008) A Myo6 mutation destroys coordination between the myosin heads, revealing new functions of myosin VI in the stereocilia of mammalian inner ear hair cells. PLoS Genet 4:e1000207
Hicks JL, Deng WM et al (1999) Class VI unconventional myosin is required for spermatogenesis in Drosophila. Mol Biol Cell 10:4341–4353
Holt JP, Bottomly K et al (2007) Assessment of myosin II, Va, VI and VIIa loss of function on endocytosis and endocytic vesicle motility in bone marrow-derived dendritic cells. Cell Motil Cytoskeleton 64:756–766
Hook V, Metz-Boutigue MH (2002) Protein trafficking to chromaffin granules and proteolytic processing within regulated secretory vesicles of neuroendocrine chromaffin cells. Ann NY Acad Sci 971:397–405
Jahn R, Lang T et al (2003) Membrane fusion. Cell 112:519–533
Jbireal JM, Strell C et al (2010) The selective role of myosin VI in lymphoid leukemia cell migration. Leuk Res 34:1656–1662
Kurz DJ, Decary S et al (2000) Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113:3613–3622
Lang T, Wacker I et al (2000) Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys J 78:2863–2877
Lister I, Schmitz S et al (2004) A monomeric myosin VI with a large working stroke. EMBO J 23:1729–1738
Majewski L, Sobczak M et al (2010) Myosin VI is associated with secretory granules and is present in nucleus in adrenal medulla chromaffin cells. Acta Biochim Polon 57:109–114
Mangold S, Wu SK et al (2011) Hepatocyte growth factor acutely perturbs actin filament anchorage at the epithelial zonula adherens. Curr Biol 21:503–507
Martin TFJ, Grishanin RN (2003) PC12 cells as a model for studies of regulated secretion in neuronal and endocrine cells. Meth Cell Biol 71:267–286
Mermall V, McNally JG et al (1994) Transport of cytoplasmic particles catalysed by an unconventional myosin in living Drosophila embryos. Nature 369:560–562
Morris SM, Arden SD et al (2002) Myosin VI binds to and localises with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic 3:331–341
Neco P, Rossetto O et al (2003) Taipoxin induces F-actin fragmentation and enhances release of catecholamines in bovine chromaffin cells. J Neurochem 85:329–337
Neco P, Giner D et al (2004) New roles of myosin II during vesicle transport and fusion in chromaffin cells. J Biol Chem 279:27450–27457
Noguchi T, Frank DJ et al (2009) Coiled-coil-mediated dimerization is not required for myosin VI to stabilize actin during spermatid individualization in Drosophila melanogaster. Mol Biol Cell 20:358–367
Osterweil E, Wells DG et al (2005) A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J Cell Biol 168:329–338
Petritsch C, Tavosanis G et al (2003) The Drosophila myosin VI Jaguar is required for basal protein targeting and correct spindle orientation in mitotic neuroblasts. Dev Cell 4:273–281
Redowicz MJ (2002) Myosins and pathology: genetics and biology. Acta Biochim Pol 49:789–804
Rose SD, Lejen T et al (2002) Molecular motors involved in chromaffin cell secretion. Ann NY Acad Sci 971:222–231
Rose SD, Lejen T et al (2003) Myosins II and V in chromaffin cells: myosin V is a chromaffin vesicle molecular motor involved in secretion. J Neurochem 85:287–298
Rudolf R, Kogel T et al (2003) Myosin Va facilitates the distribution of secretory granules in the F-actin rich cortex of PC12 cells. J Cell Sci 116:1339–1348
Sasakawa N, Ohara-Imaizumi M et al (2002) Roles of actin filaments and the actin-myosin interaction in the regulation of exocytosis in chromaffin cells. Ann NY Acad Sci 971:273–274
Spudich G, Chibalina MV et al (2007) Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat Cell Biol 9:176–183
Sweeney HL, Houdusse A (2007) What can myosin VI do in cells? Curr Opin Cell Biol 19:57–66
Trifaro JM (2002) Molecular biology of the chromaffin cell. Ann NY Acad Sci 971:11–18
Trifaró JM, Lee RW (1980) Morphological characteristics and stimulus-secretion coupling in bovine adrenal chromaffin cell cultures. Neuroscience 5:1533–1546
Vitale ML, Rodriguez del Castillo A et al (1991) Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J Cell Biol 113:1057–1067
Vitale ML, Rodríguez Del Castillo A et al (1992) Loss and Ca(2+)-dependent retention of scinderin in digitonin-permeabilized chromaffin cells: correlation with Ca(2+)-evoked catecholamine release. J Neurochem 59:1717–1728
Vreugde S, Ferrai C et al (2006) Nuclear myosin VI enhances RNA polymerase II-dependent transcription. Mol Cell 23:749–755
Wagner MC, Baryłko B et al (1992) Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol 119:163–170
Warner CL, Stewart A et al (2003) Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. EMBO J 22:569–579
Watanabe M, Nomura K et al (2005) Myosin-Va regulates exocytosis through the submicromolar Ca2+-dependent binding of syntaxin-1A. Mol Biol Cell 16:4519–4530
Wells AL, Lin AW et al (1999) Myosin VI is an actin-based motor that moves backwards. Nature 401:505–508
Wu H, Nash JE et al (2002) Interaction of SAP97 with minus-end directed actin motor protein myosin VI: implications for AMPA receptor trafficking. J Biol Chem 277:30928–30934
Yano H, Ninan I et al (2006) BDNF-mediated neurotransmission relies on upon a myosin VI motor complex. Nat Neurosci 9:1009–1018
Yoshida H, Cheng W et al (2004) Lessons from border cell migration in the Drosophila ovary: A role for myosin VI in disseminaton of human ovarian cancer. Proc Natl Acad Sci USA 101:8144–8149
Acknowledgments
We would like to thank Dr. Grazyna Mosieniak and Mr. Zbigniew Korwek for their help in flow cytometric analysis. Also, help of Prof. Monika Puzianowska-Kuznicka and Dr. Joanna Jung at the beginning of this study is greatly appreciated. This work has been supported by grant 2 P04C 037 28 and the statutory grant to the Nencki Institute from Polish Ministry of Science and Higher Education as well as by the Polish Network for Mechanisms of Cell Motility.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Majewski, Ł., Sobczak, M., Wasik, A. et al. Myosin VI in PC12 cells plays important roles in cell migration and proliferation but not in catecholamine secretion. J Muscle Res Cell Motil 32, 291–302 (2011). https://doi.org/10.1007/s10974-011-9279-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-011-9279-0