
Abstract
Sarcospan (SSPN) is a core component of the dystrophin–glycoprotein complex (DGC). Multiple SSPN transcripts are ubiquitously expressed and SSPN splicing is disrupted in many lung tumors, suggesting the importance of SSPN-related mRNAs. We describe the isolation of an alternatively spliced isoform of SSPN, which we designate ‘microspan’ based on its small size relative to SSPN. Microspan has two transmembrane domains and a novel C-terminus. We demonstrate that microspan is not an integral component of the DGC and is not perturbed by the loss of dystrophin. Microspan protein is detected at the sarcoplasmic reticulum (SR) using indirect immunofluorescence and immunoelectron microscopy. Furthermore, microspan purifies with skeletal muscle SR membranes and not transverse tubules. Mice engineered to over-express microspan display severe kyphosis and die at approximately 8 weeks of age. Levels of ryanodine receptor, dihydropyridine receptor, and SERCA-1 are greatly reduced in microspan transgenic muscle. Furthermore, electron microscopy reveals that microspan over-expression causes a dramatic perturbation in triad structure. Our findings suggest that microspan is an important component of the SR and may contribute to excitation–contraction coupling.
Similar content being viewed by others
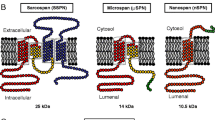
Abbreviations
- AR-LGMD:
-
autosomal recessive limb girdle muscular dystrophy
- CRUs:
-
calcium release units
- DGC:
-
dystrophin–glycoprotein complex
- DGs:
-
dystroglycans
- DHPR:
-
α1-dihydropyridine receptor
- DMD:
-
Duchenne muscular dystrophy
- EDL:
-
extensor digitorum longus
- LEL:
-
large extracellular loop
- PVDF:
-
polyvinylidene fluoride
- RyR:
-
ryanodine receptor
- SEL:
-
small extracellular loop
- SGs:
-
sarcoglycans
- SR:
-
sarcoplasmic reticulum
- SSPN:
-
sarcospan
- TA:
-
tibalis anterior
- Tg:
-
transgenic
- TM:
-
transmembrane domain
- T-tubules:
-
transverse tubules
- μSPN:
-
microspan
References
Campbell KP, Franzini-Armstrong C, Shamoo AE (1980) Further characterization of light and heavy sarcoplasmic reticulum vesicles. Identification of the ‘sarcoplasmic reticulum feet’ associated with heavy sarcoplasmic reticulum vesicles. Biochim Biophys Acta 602:97–116
Campbell KP, Kahl SD (1989) Association of dystrophin and an integral membrane glycoprotein. Nature 338:259–262
Cohn RD, Campbell KP (2000) Molecular basis of muscular dystrophies. Muscle Nerve 23:1456–1471
Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS (2000) Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol 150:1399–1410
Crosbie RH, Lim LE, Moore SA, Hirano M, Hays AP, Maybaum SW, Collin H, Dovico SA, Stolle CA, Fardeau M, Tome FM, Campbell KP (2000) Molecular and genetic characterization of sarcospan: insights into sarcoglycan–sarcospan interactions. Hum Mol Genet 9:2019–2027
Crosbie RH, Lebakken CS, Holt KH, Venzke DP, Straub V, Lee JC, Grady RM, Chamberlain JS, Sanes JR, Campbell KP (1999) Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol 145:153–165
Crosbie RH, Yamada H, Venzke DP, Lisanti MP, Campbell KP, 1998. Caveolin-3 is not an integral component of the dystrophin–glycoprotein complex. FEBS Lett 427:279–282
Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP (1997) Sarcospan, the 25-kDa transmembrane component of the dystrophin–glycoprotein complex. J Biol Chem 272:31221–31224
Ervasti JM, Campbell KP (1991) Membrane organization of the dystrophin–glycoprotein complex. Cell 66:1121–1131
Ervasti JM, Campbell KP (1993) A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122:809–823
Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345:315–319
Ervasti JM, Kahl SD, Campbell KP (1991) Purification of dystrophin from skeletal muscle. J Biol Chem 266:9161–9165
Felder E, Protasi F, Hirsch R, Franzini-Armstrong C, Allen PD (2002) Morphology and molecular composition of sarcoplasmic reticulum surface junctions in the absence of DHPR and RyR in mouse skeletal muscle. Biophys J 82:3144–3149
Heighway J, Knapp T, Boyce L, Brennand S, Field JK, Betticher DC, Ratschiller D, Gugger M, Donovan M, Lasek A, Rickert P (2002) Expression profiling of primary non-small cell lung cancer for target identification. Oncogene 21:7749–7763
Heighway J, Betticher DC, Hoban PR, Altermatt HJ, Cowen R (1996) Coamplification in tumors of KRAS2, type 2 inositol 1,4,5 triphosphate receptor gene, and a novel human gene, KRAG. Genomics 35:207–214
Holt KH, Lim LE, Straub V, Venzke DP, Duclos F, Anderson RD, Davidson BL, Campbell KP (1998) Functional rescue of the sarcoglycan complex in the BIO 14.6 hamster using delta-sarcoglycan gene transfer. Mol Cell 1: 841–848
Knudson CM, Campbell KP (1989) Albumin is a major protein component of transverse tubule vesicles isolated from skeletal muscle. J Biol Chem 264: 10795–10798
Lebakken CS, Venzke DP, Hrstka RF, Consolino CM, Faulkner JA, Williamson RA, Campbell KP (2000) Sarcospan-deficient mice maintain normal muscle function. Mol Cell Biol 20:1669–1677
Lim LE, Campbell KP (1998) The sarcoglycan complex in limb-girdle muscular dystrophy. Curr Opin Neurol 11:443–452
Michele DE, Campbell KP (2003) Dystrophin–glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem 278:15457–15460
O’Brien KF, Engle EC, Kunkel LM (2001) Analysis of human sarcospan as a candidate gene for CFEOM1. BMC Genet 2:3
Ohlendieck K, Ervasti JM, Snook JB, Campbell KP (1991) Dystrophin–glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol 112:135– 148
Spencer MJ, Guyon JR, Sorimachi H, Potts A, Richard I, Herasse M, Chamberlain J, Dalkilic I, Kunkel LM, Beckmann JS (2002) Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proc Natl Acad Sci USA 99:8874–8879
Turk, R, Sterrenburg, E, van der Wees, CG, de Meijer, EJ, de Menezes, RX, Groh, S, Campbell, KP, Noguchi, S, van Ommen, GJ, den Dunnen, JT and ‘t Hoen PA (2006) Common pathological mechanisms in mouse models for muscular dystrophies. FASEB J. 20:127–129
Yoshida M, Ozawa E (1990) Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem (Tokyo) 108:748– 752
Acknowledgements
We thank Dr. Kevin P. Campbell (HHMI, University of Iowa College of Medicine) for DHPR and RyR antibodies, Dr. Jeffrey S. Chamberlain (University of Washington, Seattle) for transgenic expression plasmids, Birgitta Sjostrand, Marianne Cilluffo and Suni Allen (Brain Research Institute electron microscopy core, UCLA) for EM experiments and Matt Schibler (Carol Moss Spivak Cell Imaging Facility, UCLA) for confocal microscopy. We are indebted to Dr. Melissa J. Spencer (UCLA) for guidance with transgene construction. A.K. Peter was supported by the Molecular, Cellular, and Integrative Physiology predoctoral training fellowship (NIH:T32GM65823). This work was supported by the Muscular Dystrophy Association (MDA3704 to G.M.), the Roy Castle Lung Cancer Foundation, UK (J.H.) and the NIH (AR48179-01 to R.H.C.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miller, G., Peter, A.K., Espinoza, E. et al. Over-expression of Microspan, a novel component of the sarcoplasmic reticulum, causes severe muscle pathology with triad abnormalities. J Muscle Res Cell Motil 27, 545–558 (2006). https://doi.org/10.1007/s10974-006-9069-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-006-9069-2