
Abstract
The complexes of N2O2-, N2O3- and N2O5-donors Schiff bases with CuII ions in the methanol solution have been synthesized. They were characterized by elemental analysis, X-ray crystallographic techniques, spectroscopic (UV–Vis, IR) and thermal (TG, TG-FTIR) methods. The catalytic activity of the prepared complexes for the hydrogen peroxide-assisted degradation of methylene blue (MB) as the model compound in water was also investigated. The complexes were obtained as crystalline solids. Depending on the Schiff bases and the molar ratio of CuII:ligand used in the synthesis, they can form monomeric or dimeric structures. The coordination environment around the metal centre in both solution and solid states is a slightly distorted square-planar. The values of magnetic moments determined at room temperature show the existence of antiferromagnetic interactions. The complexes are stable at ambient temperature. After heating, at first solvates lose solvent molecules; after that, an organic part undergoes gradual defragmentation and combustion. Degradation efficiency of MB in the presence of complexes was found to be 68.18–97.47%. A tentative mechanism involving HO· radical as an oxidant for degradation of MB was proposed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The coordination chemistry of transition metals with Schiff base has been studied extensively due to their structural diversities, versatile properties and applications [1,2,3]. Schiff base transition metal complexes have significant contribution to the development of coordination chemistry related to magnetism, catalysis and enzymatic reactions, molecular architectures and optical materials [4,5,6,7]. These complexes can also exhibit biological activity as antifungal, antibacterial, antimalarial, antitumor, antiproliferative, antiinflammatory, antiviral, antioxidant agents [8, 9]. Copper complexes attract special attention, namely due to their ability to exhibit such phenomena as molecular magnetism [10] and magnetic refrigeration [11, 12]. Several copper complexes containing Schiff base ligands have been also studied towards biological and pharmaceutical activities such as DNA binding and cleavage, antimicrobial, anticancer and antioxidant behaviour [13,14,15]. Furthermore, copper is one of the crucial transition metals which is involved in many biological processes in living organisms. The copper is required as a catalytic cofactor (e.g. cytochrome c oxidase and Cu/Zn superoxide dismutase) or as a structural component for proteins which participate in cellular reactions such as antioxidant defense, respiration, neurotransmitter biosynthesis and pigment formation. This is due to the fact that copper is highly redox active and has the ability to oxidize/reduce (switch between CuI and CuII) which allows Cu-containing proteins to play an important role as electron carriers and redox catalysts in living organisms. Copper can also have toxic effects in biological systems if not properly regulated, e.g. Menkes disease, Wilson’s disease, oxidative stress or neurodegenerative diseases [16,17,18,19,20].
The main aim of the paper was synthesis and characterization of the CuII complexes with different N,O-donor Schiff bases as well as exploration of their catalytic potential in chemical oxidation of dyes in water with hydrogen peroxide. The synthesized copper(II) complexes were characterized based on spectroscopic (IR, UV–Vis), X-ray crystallographic techniques and thermal (TG, TG-FTIR) methods as well as elemental analysis. In order to explore their catalytic properties, methylene blue (MB) was selected as a model dye because it is extensively used for dyeing and printing cotton, silk, paper, etc. [21, 22]. MB is also widely applied in different fields of medicine, e.g. as drug for tropical treatment and photodynamic therapy; as a main component of many staining solution, in antimicrobial chemotherapy or cancer research [23, 24].
Experimental
Materials
Amines (1,3-propanediamine, 2,2-dimethyl-1,3-propanediamine and 1,3-diamino-2-propanol) and ketones (2-hydroxybenzophenone and 2,2′-dihydroxybenzophenone) (Scheme 1) used for the synthesis of Schiff bases, copper(II) acetate (Cu(CH3COO)2·H2O) and solvents (MeOH, EtOH, ACN, DMF and THF) were commercially available from Sigma-Aldrich Co., Alfa Aesar Co. and Polish Chemical Reagents. All solvents and chemicals were reagent grade and used without further purification.

Synthetic route of Schiff bases synthesis
Preparation of Schiff bases
The Schiff base ligands were synthesized (Scheme 1) using a similar method described in previous papers [1, 25,26,27]. Mixtures of 10 mmol of appropriate ketone and 5 mmol of diamine in 40 mL of methanol were refluxed for 2 h. The excess of the solvent (ca. 30 mL) was then evaporated. After cooling to 4 °C, yellow solids were formed. The products were collected by filtration, washed with cold methanol and dried in air.
H 2 L 1 (where L1 = C29H24N2O2): yellow powder; yield: (62.0%); Elem. anal. for C29H26N2O2 (FW 434.53 g mol−1) (%): calcd C 80.16, H 6.03, N 6.45; found C 79.96, H 5.89, N 6.26 [25].
H 2 L 2 (where L2 = C31H28N2O2): yellow powder; yield: (74.5%); Elem. anal. for C31H30N2O2 (FW 462.58 g mol−1) (%): calcd C 80.49, H 6.54, N 6.06; found C 80.41, H 6.63; N, 5.98 [26].
H 3 L 3 (where L3 = C29H23N2O3): yellow powder; yield: (86.0%); Elem. anal. for C29H26N2O3 (FW 450.53 g mol−1) (%): calcd C 77.31, H 5.82, N 6.22; found C 76.97, H 5.75, N 6.48.
H 5 L 4·H 2 O (where L4 = C29H21N2O5): yellow powder; yield: (86.4%); Elem. anal. for C29H28N2O6 (FW 500.54 g mol−1) (%): calcd C 69.59, H 5.64, N 5.60; found C 69.26, H 5.70, N 5.68.
Synthesis of CuII complexes
The synthesis of complexes was performed in the methanol solution with the ratio of metal acetate to the Schiff base 1:1 and 2:1. An appropriate amount of copper(II) acetate dissolved in methanol (20 mL) was added to a stirred hot solution of appropriate Schiff base (0.2 g) in MeOH (30 mL). The obtained mixtures were refluxed for 1 h. The formed solid products were filtered off and washed several times with methanol. The yields of the purified complexes and some of their physicochemical properties are listed as follows:
CuL 1 (1): olive green powder; yield, 85.27%; μ eff (298 K): 1.54 μ B; Elem. anal. for CuC29H24N2O2 (FW 496,06 g mol−1) (%): calcd C 70.22, H 4.88, N 5.65; found C 69.93, H 4.84, N 5.75; FTIR bands (ν/cm−1): 3054w, 3022vw, 2963vw, 2950w, 2938w,2882vw, 2867vw, 1595m, 1578s, 1525s, 1489w, 1463m, 1454s, 1442m, 1431s, 1360w, 1340s, 1275w, 1257m, 1241s, 1180vw, 1169w, 1143s, 1119m, 1089m, 1069m, 1030m, 999w, 984w, 971w, 950m, 927vw, 908m, 871vw, 852m, 836s, 800m, 783w, 756vs, 724m, 716s, 704s, 696vs, 658m, 638m, 620w, 605vw, 595s, 586m, 544w.
CuL 2 (2): dark green powder; yield, 94.10%; μ eff (298 K): 1.55 μ B; Elem. anal. for CuC31H28N2O2 (FW 524.11 gmol−1) (%): calcd C 71.04, H 5.38, N 6.00; found C 70.78, H 5.34, N 6.01; FTIR bands (ν/cm−1) 3076vw, 3046vw, 2993vw, 2974w, 2964w, 2926vw, 2864vw, 1594m, 1569m, 1526m, 1521m, 1489w, 1467m, 1460w, 1455m, 1440m, 1429m, 1391w, 1368vw, 1355w, 1339m, 1332s, 1289w, 1273w, 1259m, 1235s, 1213w, 1180w, 1164w, 1141m, 1117w, 1073w, 1048w, 1039w, 1024w, 1000w, 977w, 969w, 952w, 925vw, 910w, 894w, 857w, 838m, 783s, 763vs, 722s, 705s, 692m, 655m, 629w, 611w, 603m, 574w, 566w, 544w.
CuHL 3 ·H 2 O (3): beige powder; yield, 67.94%; μ eff (298 K): 1.59 μ B; Elem. anal. for CuC29H26N2O4 (FW 530.07 gmol−1) (%): calcd C 65.71, H 4.94, N 5.28; found C 65.45, H 4.89, N 5.41; FTIR bands (ν/cm−1): 3647vw, 3489vw, 3053vw, 2948vw, 2922vw, 2898vw, 2834vw, 1631w, 1597s, 1582s, 1540m, 1527m, 1492w, 1464m, 1443s, 1342s, 1326w, 1314m, 1285w, 1260s, 1255s, 1247s, 1226w, 1171w, 1152m, 1119w, 1092w, 1072w, 1039vw, 1030w, 1007vw, 1001vw, 971w, 945w, 915m, 866w, 859w, 837m, 817w, 784m, 765m, 756vs, 735w, 709vs, 666w, 635m, 616m, 592m, 587m, 569vw, 548w, 541vw.
Cu 2 L 3 (CH 3 COO) (4): dark green powder; yield, 69.57%; μ eff (298 K): 2.13 μ B; Elem. anal. for Cu2C31H26N2O5 (FW 633.64 gmol−1) (%): calcd C 58.76, H 4.14, N 4.42; found C 58.75, H 3.98, N 4.52; FTIR bands (ν/cm−1): 3056vw, 3020vw, 2976vw, 2948vw, 2908vw, 2852vw, 1598m, 1579m, 1563s, 1558s, 1526s, 1490m, 1457m, 1434vs, 1410s, 1363w, 1341vs, 1289m, 1241s, 1202m, 1169w, 1147s, 1134m, 1121m, 1112m, 1087w, 1073w, 1059w, 1047w, 1029m, 1000w, 972w, 942w, 911m, 893m, 856m, 842s, 787m, 769m, 756vs, 732w, 701vs, 691s, 667s, 649m, 640s, 618m, 610m, 585w, 560w, 557w, 545w, 541w.
Cu 2 H 2 L 4 (CH 3 COO)·H 2 O·0.5CH 3 OH (5): dark turquoise powder; yield, 79.76%; μ eff (298 K): 2.18 μ B; Elem. anal. for Cu2C31.5H30N2O8.5 (FW 699.69 gmol−1) (%): calcd C 54.07′ H 4.32, N 4.00; found C 53.82, H 4.17, N 4.10; FTIR bands (ν/cm−1): 3611w, 3412vw, 3050w, 3018w, 2939w, 2858w, 2710w, 2588w, 1568m, 1537m, 1504w, 1465w, 1451s, 1434vs, 1385m, 1343m, 1321s, 1289m, 1233s, 1214s, 1170w, 1153s, 1130w, 1119m, 1099w, 1042m, 1032m, 1007w, 978w, 945w, 927w, 897w, 850s, 829m, 754vs, 728m, 707m, 688m, 668m, 644m, 629m, 614m, 562m, 548w.
Methods and physical measurements
Determination of C/H/N was done using a PerkinElmer CHN 2400 analyser. Magnetic susceptibilities for the powdered samples were measured at 298 K using a magnetic susceptibility balance MSB-MKI, Sherwood Scientific Ltd., Cambridge. The data were corrected for diamagnetic susceptibilities [28, 29]. The effective magnetic moment is calculated from Eq. (1):
An Empyrean powder diffractometer (PANalytical, the Netherlands) was used for powder X-ray diffraction (XRD) measurements. The studies were carried out on using CuKα radiation (λ = 1.5405 Å) in the 2θ range from 4° to 80° with a step of 0.026°. The X-ray powder patterns of all complexes are given in Supplementary material (Figs. S1–S5). The analytical software package WinPLOTR [30] was used to establish the positions of the peaks and to determine their intensities. Determination of the unit cell and indexing of the X-ray diffraction pattern were performed with the computer programs DICVOL06 [31]. The first twenty lines were used for the calculations, and additionally, the absolute error on each observed line was fixed at 0.03–0.06°. Reliability of the obtained unit cell was estimated by the figure of merit M N [32] and F N [33, 34]. The unit cell parameters of complexes calculated from the X-ray powder diffraction data are given in Table 1.
The FTIR spectra measurements were performed on a Nicolet 6700 FTIR using the ATR accessory on the diamond crystal for the pressed powder samples. The spectra were collected between 4000 and 540 cm−1, with a resolution of 4 cm−1 for 16 scans (Figs. S6–S10). The most important bands are given in Table 2. The UV–Vis spectra of complexes were recorded in different solvents for the solutions of compounds at a concentration 2.5 × 10−5 M; see Table 3. The measurements were performed in the 10 × 10 mm quartz cell over the range 200–1100 nm using a GENESYS 10S UV–Vis spectrophotometer. Thermal behaviour of the analysed complexes under flowing air atmosphere was studied on a Setaram Setsys 16/18 derivatograph, recording TG, DTG and DSC curves (Fig. 1; Table 4). The samples (5.2–7.6 mg) were put in a ceramic crucible and heated (10 °C min−1) in the temperature range of 30–700 °C. TG-infrared spectrometry (TG-FTIR) of the title compounds was performed using a TG Q5000 analyser interfaced to the Nicolet 6700 FTIR spectrophotometer. The appropriate complex (about 11.42–20.35 mg) was put in an open platinum crucible and heated from an ambient temperature (~23–26 °C) to 700 °C (heating rate of 20 °C min−1). The analysis was carried out under flowing nitrogen atmosphere (flow rate of 25 mL min−1). To reduce the possibility of gases condensing along the transfer line, the temperatures of the gas cell and transfer line were set to 250 and 240 °C. The FTIR spectra were recorded in the range of 4000–600 cm−1 with a resolution of 4 cm−1 and 6 scans per spectrum.

a–e TG, DTG and DSC curves of 1–5 in air; f TG curves of 1–5 in nitrogen
The crystallographic analysis was carried out using an Oxford Diffraction Xcalibur CCD diffractometer with the graphite-monochromated MoKα radiation (λ = 0.71073 Å). Data sets were collected at 293 K using the ω scan technique, with an angular scan width of 1.0°. For data collection, cell refinement and data reduction the programs CrysAlis CCD [35] and CrysAlis Red [35] were used. A multi-scan absorption correction was applied. The structures were solved by direct methods using SHELXS-2013 and refined by the full-matrix least squares on F2 using SHELXL-97 [36]; both programs are implemented in WinGX [37]. All non-H atoms were refined with the anisotropic displacement parameters. Hydrogen atoms residing on carbon atoms were positioned geometrically and refined applying the riding model [with U iso(H) = 1.2 or 1.5 U eq(C)]. The hydroxyl hydrogen did not refine satisfactorily although definitely located in a difference map, possibly due to unresolved disorder. Consequently, it was included in a calculated position (SHELXL HFIX 147 command) and as a riding contribution. The crystal data, data collection and structure refinement details are summarized in Table 5. The selected bond distances and angles are presented in Table S1 (Supplementary material). Molecular plots were drawn with ORTEP-3 for Windows [38, 39] and Mercury [40]. Geometrical calculations were performed using the PLATON program [41]. In the case of 2, the Flack parameter was refined to −0.07(5), and for further confirmation of the absolute configuration, determination using the Bayesian statistics on the Bijvoet [42] differences as implemented in the program PLATON [41] was made. The CIF file for 2 and 4 refinements can be retrieved from the Cambridge Crystallographic Data Centre (CCDC) (deposition numbers CCDC 1534459 for 2 and 1534460 for 4).
The catalytic properties of complexes towards the methylene blue dye degradation in the presence of hydrogen peroxide were determined according to the following procedure. 100 mL of MB (10 mg L−1) and 50 mg of appropriate complex were added to the conical flask and the temperature was maintained at 40 ± 2 °C. The mixtures were magnetically stirred for half hour to ensure the equilibrium was reached. Then 10 mL of 30% H2O2 was poured into solution kept continuous stirring. The reaction progress of methylene blue degradation was monitored with UV–Vis spectroscopy. The control catalysis experiment (B) was also performed under the same condition without addition of any complex.
Results and discussion
In their structure, the Schiff bases H 2 L 1 and H 2 L 2 have four possible coordination sites, two nitrogen atoms of the azomethine groups and four oxygen atoms of the phenolic groups. Therefore, the synthesis of metal complexes was conducted through the reaction between the metal ions and the ligand at 1:1 CuII:ligand ratio in methanol solutions. In the case of Schiff bases H 3 L 3 and H 5 L 4, the hydroxyl group present in the structure of amine used for the synthesis of ligands provides an additional donor atom which is capable of coordination with the copper ions. For this reason, the synthesis of complexes was carried out in the stoichiometric ratio CuII:ligand 1:1 and 2:1. For the ligand H 3 L 3, change in the ratio of the Schiff base and copper(II) ions leads to formation of two different compounds, monomer (3) and dimer (4). The analysis of the powder diffraction patterns of complexes reveals that they are crystalline compounds (see Figs S1–S5 in Supplementary material). The unit cell parameters for the studied complexes are given in Table 1. In the case of complexes 2 and 4, during the recrystallization process single crystals were obtained which allowed to perform a single-crystal X-ray analysis and verify the research results obtained by other measurements.
Infrared spectroscopy
The most characteristic bands and their assignments for the Schiff bases and their copper(II) complexes are reported in Table 2. The FTIR spectra data of CuII complexes are given in “Experimental” section and shown in Figs. S6–S10, Supplementary material. A sharp and intense peak with the maximum at 1606–1589 cm−1 on the FTIR spectra of ligands can be assigned to ν(C=N) vibration. In the complexes, this band is more intense and occurs in the same or shifted to lower frequencies indicating participation of nitrogen atoms in coordination of CuII. The metal ions are also bound by phenoxide oxygen. The O–H in-plane bending vibrations of phenolic groups are observed in the FTIR spectra of Schiff bases as the intense peaks at 1344–1297 cm−1. The lack of these bands in the spectra of the complexes can be associated with the deprotonation of the phenol groups and binding to the copper(II) ions by the oxygen atoms [26, 43, 44]. Moreover, the stretching vibration ν(C–O) occurred at 1270–1257 cm−1 in the Schiff bases spectra as a single peak; after complexation, it appears as a doublet for 1–3 and 5 [26, 43, 44] or shifts to the lower wavenumber [1, 2] which confirms coordination of metal centre by oxygen atoms of phenoxy groups. In the case of complexes 4 and 5, the bands characteristic of deprotonated carboxylic group, asymmetric and symmetric stretching vibration COO− are observed at 1563–1568 and 1432–1434 cm−1, respectively. It is noteworthy that for complex 5 the band derived from νas(COO) is coupled with ν(C=N) vibration resulting in a significant shift of the azomethine vibration towards a lower frequency compared to the free ligand.
Electronic absorption spectra and magnetic moments
The electronic spectra of complexes in protic and aprotic solvents are summarized in Table 3. The vibrations occurring below 280 nm are probably due to π → π* transitions associated with the benzene ring. These bands are more intense and split in comparison with the free ligands as a result of extensive conjugation of π electrons in the complexes indicating anionic character of the Schiff bases and the chelation of the ligands to CuII ions [3, 45]. The next band observed at 363–381 nm has a much lower molar extinction coefficient than the previous ones and generally can be assigned to the charge transfer (CT) transitions combined with π → π* transitions of the imine group [44, 46]. These CT transition bands exhibit a weak negative solvatochromism, i.e. a blue shift with the increasing solvent relative polarity. This can indicate that the ground-state molecules are better stabilized by solvation than the molecules in the excited state [25, 47, 48]. The electronic spectra of 2–5 show also bands associated with d–d transitions (2B1g→2A1g) in the range 578–631 nm. In the case of 1, the bands characteristic of the d–d transitions are not observed on the spectra at the applied concentration due to their low intensity. The low-intensity band assigned to the d–d transition appears on the spectrum of 1 recorded in more concentrated methanol solution (10−3 mol L−1) at 581 nm. Appearance of d–d transitions at such wavelengths is consistent with the square-planar geometry of CuII complexes as it was observed for other related compounds [1, 44, 49,50,51].
The μ eff values at room temperature (298 K) are 1.54–1.59 μ B for monomers 1–3 and 2.13–2.18 μ B for dimers 4 and 5. The values for complexes 1 and 2 are a bit smaller than for the expected spin-only value for uncoupled copper(II) ions (1.73 μB) which can suggest that some coupling of copper atoms can occur. The values of magnetic moments for 4 and 5 are also smaller than the expected spin-only value for two noninteracting copper(II) ions (2.45 μ B) and can be indicative of the presence of antiferromagnetic intradimer interactions [1, 52].
Thermal analysis
Thermal behaviour of complexes (1–5) was studied using the TG-DSC (air) and TG-FTIR (nitrogen) techniques. The TG-DSC curves providing information about thermal behaviour of the complexes are shown in Fig. 1.
Complex 1
As shown in Fig. 1a, complex 1 is characterized by high thermal stability. In air atmosphere, the decomposition process of 1 proceeds in two steps and starts at 283 °C. The first step is characterized by a fast mass loss rate (50.30%) and is connected with partial degradation and combustion of the Schiff base anion. The most probable product, which is formed during this stage, can be copper(II) phenolate. The theoretical mass loss value due to removal of –C17H14N2 fragment and formation of Cu(C6H5O)2 residue is 49.59%. The second stage starts at 437 °C and corresponds to degradation and combustion of the remaining part of the ligand. The final product formed at 612 °C was identified as CuO (found/calcd. total mass losses: 83.59/83.96%). The thermal decomposition of complex was also studied under nitrogen atmosphere (see Fig. 1f) together with the infrared spectroscopy analysis of gaseous decomposition products. The pyrolysis of 1 proceeds in one step, and the decomposition process is not completed under measurement conditions. The degradation process under N2 stream starts at higher temperature than it was observed in air (above 322 °C) by breaking azomethine bonds as evidenced by the presence of several bands of NH3 molecule with the characteristic maxima at 3333, 966 and 931 cm−1 at 330 °C [1, 26, 53]. Around 345 °C in the FTIR spectra of evolved gases (Fig. S11, Supplementary material), there also appear bands characteristic of CO2 (2450–2300 cm−1 and 750–600 cm−1), phenol (3649 cm−1 (ν(O–H)); 3066 cm−1 (ν(C–H)); 1607 and 1507 cm−1 (ν(Car = Car); 1339 cm−1 (δ(O–H); 1248 cm−1 (ν(C–O)); 1195 and 1177 cm−1 (δ(C–H)); 747 cm−1 (γ(C–H)); 687 cm−1 (ring)), H2O (4000–3450 and 1950–1300 cm−1) and benzonitrile (in the range 3100–3000 (ν(C–H)), 2275–2200 (ν(C≡N)) and 825–650 cm−1(δ(Car = Car), γ(CC≡N)) [1, 26, 53,54,55]. With the increasing temperature, the bands related to benzonitrile (411 °C) were not observed in the FTIR spectra, whereas several bands characteristic of toluene were recorded. The most characteristic, qualitative bands assigned to toluene occur in the ranges 3200–2800 and 800–600 cm−1 [1]. At the end of this stage in the mixture of released gases, the molecules of CO2, H2O, phenol, toluene and NH3 are found. Further heating of 1 causes slow decomposition and thermal degradation of organic part of the compound. This process is not clearly marked on the DTG curve, but the difference in mass loss recorded at 473 °C and in the final temperature (700 °C) is 2.49%. During this slow pyrolysis at 573 °C, the new gaseous products CO (double peaks at 2275–2050 cm−1 [55]) and HNCO and its methyl derivatives (3508 and 2300–2180 cm−1 [1, 53]) appear. At the end of analysis (665 °C) in the FTIR spectra of evolved gas phase, only the bands characteristic of CO, CO2, H2O, NH3, CH4, HNCO and its methyl derivatives are recorded. Probably, methane was emitted at a lower temperature, but its vibrations (several peaks at 3175–2850 cm−1 with a characteristic maximum at 3016 cm−1) were covered by the bands of phenol and toluene.
Complex 2
Complex 2 shows similar thermal stability as 1. The decomposition process of complex starts above 293 °C and proceeds in two stages. Within the first step of 2 decomposition almost 60% of initial mass loss is observed (Table 4) and an intermediate product is formed. The second stage starts above 436 °C and corresponds to combustion of the remaining part of organic ligand. The final product, CuO, is formed at 570 °C (found/calcd. overall mass loss: 85.18/84.82%). The thermal decomposition of 2 under nitrogen atmosphere undergoes also in one stage as it was observed for 1. In contrast to the previous complex, the pyrolysis starts at a similar temperature to that of the combustion process and a larger part of the ligand is degraded compared to complex 1 (more than 80%). At the beginning of pyrolysis (310 °C), the bands of CO2, H2O and aromatic compounds (probably toluene) were found in the FTIR spectra indicating that degradation of the Schiff base anion takes place in a slightly different way than observed for compound 1 (Fig. S12, Supplementary material). Breaking of the azomethine bonds starts around 346 °C; then, the vibrations of ammonia, HNCO and its methyl derivatives are recorded in FTIR spectra. With the increasing temperature (~365 °C), the bands related to benzonitrile and methane also appeared in the FTIR spectra. At the end of this stage in the FTIR spectra, only the bands characteristic of CO2, H2O, NH3, HNCO/CH3NCO, toluene and CH4 were recorded. Heating above 453 °C leads to further slow pyrolysis. Although this process is not clearly marked on the DTG curve, the difference in the mass loss at 453 °C and the final temperature of analysis (700 °C) is almost 2%. During this step, the remaining part of the unburnt fragment of organic compounds undergoes pyrolysis resulting in evolution of the following gaseous products: CO, CO2, H2O, HNCO/CH3NCO and traces amount of CH4 and toluene.
Complex 3
The TG, DTG and DSC curves corresponding to complex 3 show that decomposition under air atmosphere proceeds in three steps (Fig. 1c). The first stage of compound thermal transformation occurs at 44–120 °C and is associated with an endothermic peak which indicates that it is a desolvation process. Based on the TG-FTIR analysis, it was found that in this stage water molecules are released. A calculated mass loss due to removal of water molecule is 3.39%, while the measured mass loss is 3.14%. The enthalpy of dehydration process is equal to 29.30 kJ mol−1. In contrast to the previously described compounds, complex 3 is characterized by the lowest thermal stability, i.e. degradation and combustion of the organic part of complex begin at 218 °C. The product formed in this stage is unstable and is almost immediately further decomposed and combusted to CuO (found/calcd. total mass losses: 85.58/84.99%). The pyrolysis process of 3 occurs in two steps. The first change in the mass on the TG curve recorded in nitrogen is found to be above 44 °C; similar to the thermal decomposition of 3 in air, it is associated with the dehydration process which was confirmed by the absence of any other bands in the FTIR spectra except those related to water molecules; see Fig. 2. Further decomposition of 3 in nitrogen, linked to pyrolysis of organic ligand, is observed at a lower temperature (243 °C) compared to that of remaining complexes in this atmosphere (295–322 °C). On the basis of the FTIR spectra analysis of the evolved gas phase, it was found that water, carbon dioxide, HNCO and its methyl derivatives are released at the beginning of this process. At a slightly higher temperature (290 °C) in a mixture of the emitted gases, there are additional compounds such as ammonium and phenol. At the end of this step (above 477 °C) in the FTIR spectra, the bands characteristic of CO2, H2O, HNCO/CH3NCO and phenol were found. Similar to the previously described complexes, further heating above the final temperature of the second stage leads to slow pyrolysis of residue; the mass loss in the range of 477–700 °C is 1.5%. This part of pyrolysis is mainly related to the emission the similar gases as it was observed at the end of the second stage. Only at the end of the analysis at ~620 °C, the bands associated with phenol were not observed in the FTIR spectra of evolved volatile pyrolysis products, whereas the bands characteristic of NH3, CH4 and CO were recorded.

FTIR spectra of volatile products of thermal decomposition of 3 recorded at different temperatures
Complex 4
Complex 4 shows thermal stability comparable to that of 1 and 2. The combustion process takes place in two steps. The first change in mass accompanied by a sharp endothermic effect on the DSC curve was recorded at 293 °C. This interval corresponds to the removal and decomposition of acetic ions as well as partial combustion and transformation of Schiff base anion. Although this step is characterized by a fast mass loss rate as it was observed for the previous complexes, here a lower part of the compound is combusted; mass loss is less than 47%. The more energetic combustion process starts above 380 °C and is associated with complete degradation and oxidation of the remaining part of the organic matrix. The final product CuO is formed at 531 °C (found/calcd. overall mass loss: 75.06/74.89%).The pyrolysis process of 4 proceeds in one major step wherein the loss of about 64.53% of the initial mass is observed. At the beginning at 310 °C in the FTIR spectra, the bands characteristic of CO2, H2O, HNCO/CH3NCO and acetic acid are only recorded (Fig. S13, Supplementary material). Further heating leads to emission of ammonia (340 °C), phenol and benzonitrile (360 °C) while acetic acid disappears at 360 °C. The gaseous products, which are released and recorded on the FTIR spectra at the end of the fast stage of pyrolysis, i.e. CO2, H2O, NH3, HNCO/CH3NCO and phenol, are also observed above 531 °C where slow pyrolysis of organic matrix takes place. Additionally, around 620 °C a new gaseous product, CO, appears.
Complex 5
As shown in Fig. 1e, decomposition under air atmosphere of 5 follows five steps. The first two intervals are accompanied by endothermic effects suggesting that those are related to the desolvation process. During the first interval in the range 43–102 °C, the observed mass loss is 2.61%. This step corresponds to removal of water molecule (cald. 2.57%). The enthalpy of dehydration process is equal to 29.30 kJ mol−1. The second step of desolvation starts with an initial well-separated mass loss of 2.06%, between 110 and 194 °C and corresponding to the loss of half molecule of methanol (calcd. 2.28%). Similar processes are found on the TG curves recorded in nitrogen atmosphere. The analysis of the FTIR spectra of the evolved gas phase (Fig. 3) confirms that water molecule is a product released during the first stage, whereas during the second stage the bands characteristic of methanol and products of its partial pyrolysis (CO2 and H2O) are observed. The complex deprived of the solvents in air atmosphere is stable up to 258 °C. Heated above this temperature, it undergoes further degradation while gradual combustion of organic ligands is observed. The first stage of removal of the organic matrix is connected with removal and decomposition of acetic ions as well as partial degradation and transformation of the Schiff base anion. The formed products are unstable and immediately undergo further decomposition. In the second step of combustion, the organic part of complexes is characterized by a significant reduction in the mass loss rate (5.07%). The remaining of the Schiff base is oxidative eliminated in the last step with CuO formation; the found and calculated total mass losses are 77.58 and 77.26%, respectively. In nitrogen atmosphere after the desolvation process, further thermal decomposition of 5 proceeds in two steps and it can be observed from the TG curve that the decomposition process was not completed under the measurement condition (Fig. 1f). The first step (309–407 °C) is assigned to removal of acetate ion as well as destruction and partial pyrolysis of the Schiff base. This is confirmed by the FTIR spectra analysis of the evolved gas phase. At 315 °C, the bands of acetic acid, CO2, H2O and HNCO/CH3NCO are observed on the spectra of emitted gases. The bands characteristic of ammonia, benzonitrile and phenol appear around 325, 340 and 345 °C, respectively, confirming further degradation of the Schiff base. Before the beginning of the second stage of decomposition process, slow pyrolysis, which is not clearly marked on the DTG curve, is observed. The mass loss between 407 and 581 °C is ~3.7%. The main gases during this interval are CO2, H2O, NH3 and HNCO/CH3NCO. The last stage of pyrolysis is associated with emission of gases similar to those mentioned above as well as CO and CH4.

FTIR spectra of volatile products of thermal decomposition of 5 recorded at different temperatures
Structure of 2 and 4 complexes
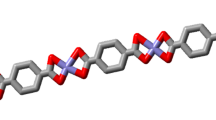
During recrystallization of complexes 2 and 4 from methanol solutions, the single crystals were obtained which made it possible to carry out a single-crystal X-ray analysis and confirm the results of other studies. The molecular structures of complexes 2 and 4 are given in Fig. 4. The crystallographic and refinement data are summarized in Table 5. The selected bond distances and angles are given in Table S1, and the hydrogen bond parameters are listed in Table S2 (see Supplementary material). The similar structure of complex 2 but obtained from the ethanol solution was reported earlier by Kargar et al. [56]. For this reason, it is not discussed here in detail. The results of the measurements are given in order to confirm the correctness of determination of the unit cell parameters calculated based on the data obtained from the powder diffraction analysis as well as the coordination around the metal ion. CuII ion is bound by oxygen atoms of the deprotonated phenolic groups as well as two imine nitrogen atoms. The structural index parameter τ 4 is 0.45 which can indicate that the coordination environment around CuII ion is a very distorted square-planar geometry; the values of τ 4 range from zero (ideal square-planar geometry) to 1.00 (perfect tetrahedral geometry) [57].

ORTEP view with the atom numbering scheme of 2 and 4. Displacement ellipsoids are drawn at the 50% probability level
In the case of complex 4, during the recrystallization process the methanol molecule is incorporated into the structure of the compound and affected the unit cell parameters which are slightly different from that calculated for the solvent-free compound (Table 1). Complex 4 crystallizes in the centrosymmetric triclinic space group P \(\bar{1}\) as a dimer. As shown in Fig. 4, the deprotonated ligand, L3−, acts as a hexadentate one. The complex is dinuclear having metal centres doubly bridged by the μ-alkoxo oxygen and the μ-acetate anion giving rise to a Cu2O3 metallacycle with the Cu1–Cu2 separation of 3.468(1) Å and the Cu1–O2–Cu2 angle equals 132.05(13)°. This distance is in the range of similar dinuclear copper(II) complexes [58,59,60]. According to the literature [58,59,60,61,62], the μ-oxo bridged CuII complexes are usually antiferromagnetic with the Cu–O–Cu angle being greater than 120–135° and the larger angles should lead to stronger antiferromagnetic coupling. The existence of some intramolecular antiferromagnetic interactions in the studied complex was confirmed by the magnetic susceptibility measurement at 298 K. In addition, each CuII ion is coordinated to one imine nitrogen and one phenoxo oxygen originating from ligand. The square-planar NO3 coordination geometry around both CuII ions is slightly tetrahedrally distorted as indicated by structural index parameters τ4 which are 0.05 for Cu1 and 0.11 for Cu2. The higher tetrahedral distortion of geometry around Cu2 ion is also apparent from the dihedral angles (θ = 11.00°) between two N–Cu–Ophenoxo and Ocarboxylate–Cu–Oalkoxo planes, whereas for Cu1 it equals 5.27°. The \(\text{NO}_{3}{-}\) core around Cu1 is nearly planar where the largest deviation is 0.026(1) Å for N1 atom; the Cu1II ion lies in the mentioned plane with the deviation being −0.056(1) Å. The RMS deviation of the four coordinating atoms (\(\text{NO}_{3}{-}\) core) of Cu2II ion from their best plane is 0.129 Å; the Cu2II ion lies almost in the mentioned plane with the deviation being only 0.031(1) Å. The dihedral angle between the two coordination planes is 15.63(8)° indicating that the bridging moiety differs slightly from planarity. The Cu–O (1.879(2)–1.944(2) Å) and Cu–N (1.951(3), 1.941(3) Å) bond lengths are similar to the corresponding distances observed in the other Schiff base CuII complexes of square-planar coordination [44, 60, 63].
Methanol molecule is linked to the complex via the intermolecular three-centre bifurcated intramolecular hydrogen bond with two oxygen atoms of phenoxy and carboxylate groups. The structure is also stabilized by C–H···O hydrogen bonds and C–H···π interactions. The presence of weak C–H···O hydrogen bond results in the formation of a one-dimensional network; see Fig. 5. Such 1D arrays were further packed in the parallel fashion stabilized by the C–H···Cg interactions leading to formation of a 2D network (Figure S14, Supplementary material).

Part of the crystal structure of 4 showing formation of 1D polymer along the a-axis
Catalytic properties
Copper(II) is an established Fenton catalyst that can react with oxygen or peroxide to generate highly reactive oxygen species (ROS). In order to study the catalytic ability of synthesized complexes for the degradation of organic dyes, MB was used as a model pollutant in the aqueous media due to its highly stable structure and being easy to monitor. The degradation of methylene blue with H2O2 in the presence of complexes was monitored by UV–Vis spectroscopy at 665 nm through the discoloration of the dye solution. The concentration of MB solution was determined quantitatively through the calibration graph constructed from the solutions of MB at various concentrations (R 2 = 0.9988). The discoloration ratio (η) was calculated from Eq. (2):
where c t and c o represent the time-dependent and initial concentrations of dye (mg L−1), respectively.
The discoloration results are shown in Fig. 6. Significant catalytic properties towards degradation of MB are exhibited by the complexes of Schiff bases synthesized from 1,3-diamino-2-propanol. As shown in Fig. 6, complex 3 acts very fast. After half an hour, more than 15% of MB undergoes degradation. This can be due to the complex form, i.e. fine crystalline powder, which is dispersed while stirring in the whole volume of solution forming a suspension. Compound 4 shows the most effective catalytic properties. This complex did not react so quickly with hydrogen peroxide as 3, but after 4 h more than 80% of MB was decomposed. The good catalytic activity is also observed in the case of complex 5. After 7 h, more than 97% of dye was oxidized. In the case of 4 and 5, effective catalytic properties can be associated with their structure, i.e. the fact that these are the dimers. Moreover, from the FTIR spectra (Fig. S15, Supplementary material) it can be clearly seen there are no changes in characteristic peaks of complexes and discoloration process is due to catalytic oxidation of MB than adsorption on the complexes surface. Complexes 1 and 2 show a lower degradation ability of dye than the remaining compounds; degradation efficiency of MB is around 70% after 7 h. Catalytic properties of the studied complexes towards degradation of MB with H2O2 can be explained by a mechanism similar to those reported by dos Santos et al. [64] and Shan et al. [65]. CuII ions can decompose hydrogen peroxide, reducing to CuI and generating the hydroperoxyl radical (HOO•) and the superoxide anion (•\({\text{O}}_{2}^{ - }\)):
Then, the cuprous ions can react with H2O2 and generate the most reactive ROS, i.e. the hydroxyl radical which can be responsible for direct oxidation and degradation MB:
In the blank solution, a decrease in the concentration of MB is also observed. The discoloration can be explained by formation of •OH radical during homolysis of hydrogen peroxides induced by light as well as heat [66, 67]. In comparison with the solutions containing the complexes addition, this process is slower; a significant change in the concentration of MB (<2%) can be seen only after an hour.

Discoloration plot of methylene blue oxidation in the presence of H2O2 and CuII complexes as catalysts
Conclusions
The complexes of N2O2-, N2O3- and N2O5-donors Schiff bases with CuII ions in the methanol solution were synthesized and characterized using different physiochemical methods. Depending on the Schiff bases used in the synthesis and the molar ratio of CuII:ligand, they can form monomeric or dimeric structures. The coordination environment around the metal centre in both solution and solid states is a slightly distorted square planar. The complexes are stable at room temperature. In the case of complexes 3 and 5, thermal decomposition of compounds is preceded by the desolvation process. The TG-FTIR analysis confirms the presence of water (for 3 and 5) and methanol (for 5) molecules in their structures. Then, in the case of 4 and 5, the acetate ions are removed from the structures of complexes, which is confirmed by the presence of acetic acid bands in the FTIR spectra of gaseous products. The thermal degradation of the Schiff bases occur in consecutive reactions where in almost all complexes (except 2) azomethine double bonds firstly undergo the destruction which is confirmed by the TG-FTIR analysis; the ammonia and HNCO/CH3NCO bands appear as the first on the FTIR spectra of evolved gases. Heating in nitrogen also resulted in evolution of: CO2, CO, water, methane, toluene/phenol and benzonitrile molecules as volatile products of organic part thermal decomposition. The complexes exhibit catalytic properties towards oxidation of MB in water with hydrogen peroxide. A tentative mechanism involving the HO· radical as an oxidant for degradation of MB was reported.
References
Bartyzel A. Synthesis, thermal study and some properties of N2O4-donor Schiff base and its Mn(III), Co(II), Ni(II), Cu(II) and Zn(II) complexes. J Therm Anal Calorim. 2017;127:2133–47.
Cristóvão B, Miroslaw B. Propeller-like heterotrinuclear CuII–LnIII–CuII compounds—physicochemical properties. Inorg Chim Acta. 2013;401:50–7.
Shafaatian B, Mousavi SS, Afshari S. Synthesis, characterization, spectroscopic and theoretical studies of new zinc(II), copper(II) and nickel(II) complexes based on imine ligand containing 2-aminothiophenol moiety. J Mol Struc. 2016;1123:191–8.
Liua K, Shia W, Chenga P. Toward heterometallic single-molecule magnets: synthetic strategy, structures and properties of 3d–4f discrete complexes. Coord Chem Rev. 2015;289–290:74–122.
Pradeep CP, Das SK. Coordination and supramolecular aspects of the metal complexes of chiral N-salicyl-β-amino alcohol Schiff base ligands: towards understanding the roles of weak interactions in their catalytic reactions. Coord Chem Rev. 2013;257:1699–715.
Gupta KC, Sutar AK. Catalytic activities of Schiff base transition metal complexes. Coord Chem Rev. 2008;252:1420–50.
Yang X, Jones RA, Huang S. Luminescent 4f and d-4f polynuclear complexes and coordination polymers with flexible salen-type ligands. Coord Chem Rev. 2014;273–274:63–75.
Kumar S, Dhar DN, Saxena PN, Kanpur IIT. Applications of metal complexes of Schiff bases—a review. J Sci Ind Res. 2009;68:181–7.
Bartyzel A, Cukrowska EM. Solid phase extraction method for the separation and determination of chromium(III) in the presence of chromium(VI) using silica gel modified by N, N’-bis-(alpha-methylsalicylidene)-2,2-dimethyl-1,3-propanediimine. Anal Chim Acta. 2011;707:204–9.
Ardizzoia GA, Brenna S. Hydroxo-bridged copper(II) cubane complexes. Coord Chem Rev. 2016;311:53–74.
Miao XH, Han SD, Liu SJ, Bu XH. Two lanthanide(III)–copper(II) chains based on [Cu2Ln2] clusters exhibiting high stability, magnetocaloric effect and slow magnetic relaxation. Chin Chem Lett. 2014;25:829–34.
Happ P, Plenk C, Rentschler E. 12-MC-4 metallacrowns as versatile tools for SMM research. Coord Chem Rev. 2015;289–290:238–60.
Reddy PR, Rajeshwar S, Satyanarayana B. Synthesis, characterization of new copper(II) Schiff base and 1,10-phenanthroline complexes and study of their bioproperties. J Photochem Photobiol B Biol. 2016;160:217–24.
Chakraborty A, Kumar P, Ghosh K, Roy P. Evaluation of a Schiff base copper complex compound as potent anticancer molecule with multiple targets of action. Eur J Pharm. 2010;647:1–12.
Kostova I, Saso L. Advances in research of Schiff-base metal complexes as potent antioxidants. Curr Med Chem. 2013;20:4609–32.
Park KC, Fouani L, Jansson PJ, Wooi D, Sahni S, Lane DJR, Palanimuthu D, Lok HC, Kovačević Z, Huang MLH, Kalinowski DS, Richardson DR. Copper and conquer: copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics. 2016;8:874–86.
Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Aspects Med. 2005;26:268–98.
Cerpa W, Varela-Nallar L, Reyes AE, Minniti AN, Inestrosa NC. Is there a role for copper in neurodegenerative diseases? Mol Aspects Med. 2005;26:405–20.
Harris ED. Basic and clinical aspects of copper. Crit Rev Cl Lab Sci. 2003;40:547–86.
Tapiero H, Townsend DM, Tew K. Trace elements in human physiology and pathology.Copper. Biomed Pharmacother. 2003;57:386–98.
Wu EMY, Kuo SL. Decolourization of methylene blue in water using bentonite impregnated with Ti and Ag as photocatalyst. Water Environ Res. 2015;87:727–34.
Chen L, Li Y, Du Q, Wang Z, Xia Y, Yedinak E, Lou J, Ci L. High performance agar/graphene oxide composite aerogel for methylene blue removal. Carbohyd Polym. 2017;155:345–53.
Orth K, Beck G, Genze F, Rück A. Methylene blue mediated photodynamic therapy in experimental colorectal tumors in mice. J Photochem Photobiol, B. 2000;57:186–92.
Albadarin AB, Collins MN, Naushad M, Shirazian S, Walker G, Mangwandi C. Activated lignin-chitosan extruded blends for efficient adsorption of methylene blue. Chem Eng J. 2017;307:264–72.
Bartyzel A. Effect of solvents on synthesis and recrystallization of Ni(II) complex with N2O2-donor Schiff base. Inorg Chim Acta. 2017;459:103–12.
Bartyzel A, Kaczor AA. The formation of a neutral manganese(III) complex containing a tetradentate Schiff base and a ketone—synthesis and characterization. J Coord Chem. 2015;68:3701–17.
Black R, Billing D, Bartyzel A, Cukrowska E. 2,2′-[(Propane-1,3-diyldinitrilo)bis(phenylmethylidyne)]diphenol. Acta Crystallogr. 2010;E66:o1002-u3755.
Bain GA, Berry JF. Diamagnetic corrections and Pascal’s constants. J Chem Educ. 2008;85:532.
Carlin RL. Magnetochemistry. Heidelberg: Springer; 1986.
Roisnel T, Rodriguez-Carvajal J. In: Delhez R, Mittenmeijer EJ, editors. Proceedings of the seventh European powder diffraction conference (EPDIC 7), 2000. p. 118–23.
Boultif A, Louër D. Powder pattern indexing with the dichotomy method. J Appl Cryst. 2004;37:724–32.
de Wolff PM. A simplified criterion for the reliability of a powder pattern indexing. J Appl Cryst. 1968;1:108–13.
Smith GS, Snyder RL. FN: a criterion for rating powder diffraction patterns and evaluating the reliability of powder-pattern indexing. J Appl Cryst. 1979;12:60–5.
Boultif A, Louër D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J Appl Cryst. 1991;24:987–93.
CrysAlis PRO, editor. Yarnton. Oxfordshire: UK Agilent Technologies Ltd.; 2013.
Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A. 2008;64:112–22.
Farrugia LJ. WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr. 1999;32:837–8.
Farrugia LJ. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J Appl Cryst. 1997;30:565.
Farrugia LJ. WinGX and ORTEP for Windows: an update. J Appl Cryst. 2012;45:849–54.
Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. Mercury: visualization and analysis of crystal structures. J Appl Cryst. 2006;39:453–7.
Spek A. Single-crystal structure validation with the program PLATON. J Appl Cryst. 2003;36:7–13.
Hooft RWW, Straver LH, Spek AL. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J Appl Crystallogr. 2008;41:96–103.
Cristóvão B, Kłak J, Miroslaw B. Synthesis and characterization of CuII–LnIII (Ln = Ho, Tm, Yb, or Lu) complexes with N2O4-donor Schiff base ligand. J Coord Chem. 2014;67:2728–46.
Bartyzel A, Głuchowska H. Influence of temperature on the crystallization of CuII complex with tetradentate Schiff base. J Coord Chem. 2016;69:3206–18.
Grivani G, Vakili M, Khalaji AD, Bruno G, Rudbari HA, Taghavi M. Schiff bases synthesis, characterization, crystal structure determination and computational study of a new Cu(II) complex of bis [2-{(E)-[2-chloroethyl)imino]methyl}phenolato)] copper(II) Schiff base complex. J Mol Struct. 2016;1116:333–9.
Fraser C, Bosnich B. Bimetallic reactivity. Investigation of metal-metal interaction in complexes of a chiral macrocyclic binucleating ligand bearing 6- and 4-coordinate sites. Inorg Chem. 1994;33:338–46.
Reichardt C. Solvatochromic dyes as solvent polarity indicators. Chem Rev. 1994;94:2319–58.
Trujillo A, Fuentealba M, Carrillo D, Manzur C, Hamon JR. Synthesis, characterization and X-ray crystal structure of an allyloxo functionalized nonsymmetric nickel coordination complex based on N2O2 chelating ferrocenyl ligand. J Organomet Chem. 2009;694:1435–40.
John A, Katiyar V, Pang K, Shaikh MM, Nanavati H, Ghosh P. Ni(II) and Cu(II) complexes of phenoxy-ketimine ligands: synthesis, structures and their utility in bulk ring-opening polymerization (ROP) of l-lactide. Polyhedron. 2007;26:4033–44.
Shakir M, Hanif S, Sherwani MA, Mohammad O, Al-Resayes SI. Pharmacologically significant complexes of Mn(II), Co(II), Ni(II), Cu(II) and Zn(II) of novel Schiff base ligand, (E)-N-(furan-2-yl methylene) quinolin-8-amine: synthesis, spectral, XRD, SEM, antimicrobial, antioxidant and in vitro cytotoxic studies. J Mol Struct. 2015;1092:143–59.
Das M, Ghosh BN, Valkonen A, Rissanen K, Chattopadhyay S. Copper(II) complexes with tridentate N2O donor Schiff base isomers: modulation of molecular and crystalline architectures through supramolecular interactions. J Saudi Chem Soc. 2013;17:269–76.
Ismayilov RH, Wang WZ, Lee GH, Peng SM. One-, two- and three-dimensional Cu(II) complexes built via new oligopyrazinediamine ligands: from antiferromagnetic to ferromagnetic coupling. Dalton Trans. 2006:478–91.
Bartyzel A, Sztanke M, Sztanke K. Thermal studies of analgesic active 8-aryl-2,6,7,8-tetrahydroimidazo[2,1-c][1, 2, 4]triazine-3,4-diones. J Therm Anal Calorim. 2016;123:2053–60.
Zong G, Ren N, Zhang J, Qi X, Gao J. Lanthanide complexes with 3-bromine-4-methyl benzoic acid and 1,10-phenanthroline. J Therm Anal Calorim. 2016;123:105–16.
Bartyzel A. Synthesis, crystal structure and characterization of manganese(III) complex containing a tetradentate Schiff base. J Coord Chem. 2013;66:4292–303.
Kargar H, Kia R, Moghadam M, Froozandeha F, Tahir MN. {2,2′-[(2,2-Dimethylpropane-1,3-diyldinitrilo)bis(phenylmethylidyne)]diphenolato}copper(II). Acta Cryst. 2011;E67:m1145.
Yang L, Powell DR, Houser RP. Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans 2007:955–64.
Kavlakoglu E, Elmali A, Elerman Y. Magnetic super-exchange mechanism and crystal structure of a binuclear μ-acetato-bridged copper(II) complex of pentadentate binucleating ligand. An influence of overlap interactions to magnetic properties. Z Naturforsch, B: Chem Sci. 2002;57:323–30.
Mukherjee A, Saha MK, Nethaji M, Chakravarty AR. Dicopper(II) Schiff base aminobenzoates with discrete molecular and 1D-chain polymeric structures. Polyhedron. 2004;23:2177–82.
Elmali A, Zeyrek CT, Elerman Y. Crystal structure, magnetic properties and molecular orbital calculations of a binuclear copper(II) complex bridged by an alkoxo-oxygen atom and an acetate ion. J Mol Struct. 2004;693:225–34.
Negodaev I, de Graaf C, Caballol R, Lukov VV. On the magnetic coupling in asymmetric bridged Cu(II) dinuclear complexes: the influence of substitutions on the carboxylato group. Inorg Chim Acta. 2011;375:166–72.
Weng CH, Cheng SC, Wei HM, Wei HH, Lee CJ. Magnetostructural correlations and catecholase-like activities of l-alkoxo-l-carboxylato double bridged dinuclear and tetranuclear copper(II) complexes. Inorg Chim Acta. 2006;359:2029–40.
Nishida Y, Kida S. Crystal structures and magnetism of binuclear copper(II) complexes with alkoxide bridges. Importance of orbital complementarity in spin coupling through two different bridging groups. J Chem Soc Dalton Trans. 1986:2633–40.
dos Santos PL, Guimarães IR, Mesquita AM, Guerreiro MC. Copper-doped akaganeite: application in catalytic Cupro-Fenton reactions for oxidation of methylene blue. J Mol Catal A: Chem. 2016;424:194–202.
Shan Z, Lu M, Wang L, MacDonald B, MacInnis J, Mkandawire M, Zhang X, Oakesaf KD. Chloride accelerated Fenton chemistry for the ultrasensitive and selective colorimetric detection of copper. Chem Commun. 2016;52:2087–90.
Feuerstein O, Moreinos D, Steinberg D. Synergic antibacterial effect between visible light and hydrogen peroxide on Streptococcus mutans. J Antimicrob Chemother. 2006;57:872–6.
Gligorovski S, Strekowski R, Barbati S, Vione D. Environmental implications of hydroxyl radicals (·OH). Chem Rev. 2015;115:13051–92.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bartyzel, A. Synthesis, thermal behaviour and some properties of CuII complexes with N,O-donor Schiff bases. J Therm Anal Calorim 131, 1221–1236 (2018). https://doi.org/10.1007/s10973-017-6563-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6563-2