
Abstract
New phase of a formula Fe0.83V1.17O4 (Fe1−xV1+xO4, x = 0.17) has been obtained by quenching molten mixture comprising 75 mol% of V2O5 and 25 mol% of α-Fe2O3 and subsequent leaching the excessive solid solution of Fe2O3 in V2O5 with the help of 6 mol solution of NaOH. New phase crystallizes in a monoclinic system with unit cell parameters: a = 0.9914(3) nm, b = 0.8967(2) nm, c = 0.6852(2) nm, β = 107.69(4)°. It is isostructural with α-MnMoO4. It was found that at 670 °C this phase undergoes a transition to phase of FeVO4-I structure. IR spectrum of the Fe0.83V1.17O4 has been recorded.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transition metal orthovanadates of general formula MVO4, where M = Cr, Fe, In, have been extensively studied because of their potential use as electrode materials in lithium secondary batteries [1–4]. One of the most attractive properties of orthovanadates during the insertion of lithium ions is their high charge insertion capacity. The subject of investigations of numerous works are thin films containing amorphous, nanocrystaline, or well-crystallized orthovanadates [2, 4]. Among others, iron(III) orthovanadate(V) is one of the most frequently investigated [1–4]. The survey of publications has shown that Fe2V4O13, FeVO4, and solid solution of the Fe2O3 in V2O5, with solubility limit reaching 3 mol% of Fe2O3, are formed in the two-component system of Fe2O3–V2O5 [5–13]. The basic properties of Fe2V4O13 and FeVO4 are known. The phase Fe2V4O13 crystallizes in a monoclinic system, space group P21/C [5]. It melts incongruently at 665 °C with deposition of FeVO4 [6]. On the other hand, there are known four polymorphs of iron(III) orthovanadate(V) [5–12]. FeVO4-I, crystallizes in a triclinic system (S.G. P-1) [7, 8] and is stable at ambient conditions [6–11]. FeVO4-I melts incongruently at 850 °C with deposition of α-Fe2O3 as a solid product of melting [6]. Three other metastable polymorphs, the orthorhombic FeVO4-II, isostructural with CrVO4-III [9], the orthorhombic FeVO4-III adopting α-PbO2 structure [10] and monoclinic, wolframite structure FeVO4-IV [11] are normally formed under high pressure and high temperature conditions [6–12]. According to Muller and Joubert [11], FeVO4-III and FeVO4-IV were obtained directly by the high pressure, high temperature synthesis. On the other hand, orthorhombic FeVO4-II was obtained from FeVO4-III around 540 °C, and has narrow range of stability, reaching 570 °C [11]. The metastable FeVO4-II can be obtained also by hydrothermal method [9]. Muller and Joubert [11] suggest additionally that FeVO4-III is a reduced form of composition Fe1−xV1+xO4 containing some tetravalent vanadium. The results of thermogravimetric (TG) investigations have shown that FeVO4 prepared using solution method corresponds to amorphous phase of the FeVO4·2.7H2O formula [3]. The X-ray data of the amorphous phase annealed at 500 °C show the beginning of crystallization, yielding well-crystallized triclinic FeVO4-I after annealing at 600 °C. On the other hand, the studies conducted with the help of transmission electron microscopy and focused on preparation of the FeVO4 thin films have revealed that amorphous iron(III) orthovanadate(V) after thermal treatment at 300 °C transforms to orthorhombic FeVO4-II, and next after sintering at 500 °C it transforms to triclinic FeVO4-I [2].
The literature survey has shown as well that in the FeVO4–CrVO4 system solid solution of general formula Fe1−xCrxVO4, for 0.1 < x < 0.85, forms, which adopts α-MnMoO4 structure [13, 14]. Thus, the structure of the Fe1−xCrxVO4 phase differs essentially from the stable at ambient conditions structures of triclinic FeVO4-I and orthorhombic CrVO4-III. On the other hand, soft chemistry synthesis methods lead to formation of CrVO4-I and InVO4-I isostructural with α-MnMoO4 [15, 16].
Despite numerous works concerning the Fe2O3–V2O5 system, the investigations conducted lately in Department of Inorganic and Analytical Chemistry and focused on verification of the literature data concerning properties of FeVO4 have revealed the presence of new phase in its melting products. Powder diffraction patterns of molten and quenched samples are frequently very complex and contain diffraction lines which are shifted, broaden, overlapped, and weak. It makes identification of single phases in such samples very difficult. Despite potential problems, the aim of this study was to obtain this new phase, denoted as X phase, and to investigate its properties with the help of XRD, DTA/TG, SEM/EDX, and IR methods. Results of investigations by XRD, DTA/TG, SEM/EDX, and IR methods allow a determination of composition of samples and establishing the range of their thermal stability [17–20].
Experimental
The materials used in the research were V2O5, p.a. (POCh, Poland), α-Fe2O3 p.a. (POCh, Poland), NaOH p.a. (POCh, Poland), and made in-house FeVO4-I. FeVO4-I was synthesized from stoichiometric mixture of oxides in the following stages: 560 °C (20 h) + 590 °C (20 h) × 2 [17, 18].
The DTA/TG examinations were made using an apparatus of Paulik–Paulik–Erdey type (MOM, Hungary). Samples of 500 mg were investigated in air up to the 1,000 °C in quartz crucibles and at the heating rate of 10 °C min−1.
X-ray phase analysis (XRD) of the samples was performed using a HZG-4 diffractometer (Carl Zeiss, Germany) applying the Co Kα/Fe radiation (step 0.02° 2θ, time 1 s). The identification of the individual phases was performed on the ground of their characteristics contained in PDF cards. The powder diffraction pattern of the new phase was indexed with the help of the Refinement program of DHN/PDS package, using α-Al2O3 as the internal standard.
The density of the new phase was determined in argon with the help of the Ultrapyc 1200e ultrapycnometer (Quantachrome Instruments, USA).
The investigations by SEM/EDX methods were carried out by using of an electron scanning microscope (JSM-1600, Jeol, Japan) matched with an X-ray energy dispersive spectrometer (ISIS-300, Oxford Instruments).
The investigations with made in-house FeVO4-I were conducted using fine ground samples subjected to heat treatment in different conditions. For the further experiments were selected five samples containing 10, 25, 33, 67, and 75 mol% of Fe2O3 in mixture with V2O5. The oxides weighted in an appropriate proportions after grinding in an agate mortar were heated at 900 °C for 3 h, quenched to room temperature, and subjected to X-ray phase analysis (XRD).
The single phase sample of the new phase X was obtained using mixture of oxides containing 25 mol% of α-Fe2O3 and 75 mol% of V2O5. A mixture of 7.736 g of V2O5 and 2.264 g of α-Fe2O3 was heated in crucible at 900 °C for 3 h and quenched to room temperature yielding a brown solid. After fine grinding in agate mortar, the excessive amounts of V2O5 were removed from solidified melt by leaching with 100 ml of 6 mol solution of NaOH. Next, the obtained solid was filtered using paper filter, washed several times with distilled water and dried at 110 °C for 4 h in drying oven. The phase composition of prepared phase was controlled with the help of XRD method. The dark brown solid obtained according to this procedure, the phase X, was subjected to further investigations.
Results and discussion
In order to obtain new phase, denoted as X phase, two series of experiments were conducted. In the first stage of research, experiments were concerning the heat treatment of FeVO4 in different conditions. At the beginning, the sample of FeVO4 was subjected to investigation with the help of DTA method. On the DTA curve of FeVO4 one endothermic effect was recorded, with onset temperature at 850 °C, and maximum temperature at 900 °C, attributed to melting of this phase. These temperatures are in accord with the literature data [6]. In order to know the products of FeVO4 melting, sample of FeVO4 was heated for 3 h at 900 °C that is at temperature of maximum of endothermic effect attributed to its melting, and quenched to room temperature. The results of the X-ray phase analysis of this sample after heat treatment (Table 1, no. 2) have shown that besides V2O5 and α-Fe2O3 this sample contains phase X. The diffraction lines characteristic for V2O5 recorded on powder diffraction pattern of this sample were shifted slightly towards higher 2θ angles. As described in the literature, it indicated formation of solid solution of Fe2O3 in V2O5 [6], denoted in this work as a V2O5(ss). Moreover, at 900 °C V2O5 does not exists as a solid, thus it crystallizes from liquid, and cannot be treated as a solid product of melting.
The results of a X-ray qualitative phase analysis of FeVO4 samples heat treated in different conditions are given in Table 1. Analysis of these data permits advancing suggestion, in order to obtain phase X, samples have to be heated at temperatures higher than the temperature of melting of FeVO4 (at 900 or 1,000 °C) and quenched to room temperature. Based on these conclusions, in the second stage of investigations samples were heated at 900 °C for 3 h and quenched to room temperature.
In the second stage of research, several mixtures comprising V2O5 and α-Fe2O3 were heated at 900 °C for 3 h and quenched to room temperature. The compositions of samples and results of X-ray qualitative phase analysis after heat treatment are given in Table 2.
The measurement results shown in Table 2 have given rise to statement that only sample of initial composition of 75 mol% of V2O5 and 25 mol% of Fe2O3 after quenching contains a new phase X and solid solution of Fe2O3 in V2O5. This solid solution was removed from sample by leaching with 6 mol. NaOH solution yielding pure phase X. The powder diffraction pattern of the X phase was subjected to indexing of which results are given in Table 3. Phase X crystallizes in the monoclinic system and its end-centered (C) unit cell parameters are given in Table 4. The literature scan has shown that angular positions of diffraction reflection, their relative intensities and unit cell parameter of phase X are similar to these ones of Fe1−x Cr x VO4 solid solution phase, CrVO4-I and InVO4-I, which are isostructural with α-MnMoO4 [13–16]. It permits advancing suggestion, that phase X belongs to a group of orthovanadates, MVO4, isostructural with α-MnMoO4.
The sample of initial composition of 75 mol% of V2O5 and 25 mol% of Fe2O3 heated at 900 °C for 3 h and quenched to room temperature and containing a mixture of a new phase X and solid solution of Fe2O3 in V2O5 was subjected to investigations with the help of an electron scanning microscope matched with an X-ray energy dispersive spectrometer. A SEM image has revealed that in the surface layer of this sample needle-like crystals are prevailing, with their lengths confined in the range of 0.2–1 mm (Fig. 1). The crystallites from deeper layers were smaller and more irregular in shape (Fig. 1). The results of experimental determination of composition by EDX analysis of needle-like crystals showed that the Fe:V ratio is near to 10:14 (on average: 41.52 at.% Fe and 58.48 at.% V) and it corresponds to phase X. Taking under consideration orthovanadate formula MVO4, it gives Fe0.83V1.17O4 (Fe1−xV1+xO4, for x = 0.17). It resembles the case of FeVO4-III, where the partial reduction of vanadium ions leads to formula Fe1−xV1+xO4 [11].

A SEM image of a sample of initial composition of 75 mol% of V2O5 and 25 mol% of Fe2O3 heated at 900 °C for 3 h and quenched to room temperature. Magnification ×50
On the other hand, the results of experimental determination of composition of crystallites from deeper layers have revealed the presence of two phases: phase X and solid solution of Fe2O3 in V2O5. The results of experimental determination of composition of solid solution showed that the V:Fe ratio is close to 24:1 (on average 96.16 at.% V and 3.84 at.% Fe) and is in accord with the literature data [6].
The measured density of Fe0.83V1.17O4 amounts to d exp = 3.94(5) g cm−3, and is in accord with XRD density d c = 3.89 g cm−3 calculated for eight asymmetric units in the unit cell (Z = 8, characteristic for α-MnMoO4 structure).
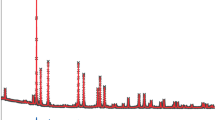
The sample of pure phase Fe0.83V1.17O4 was additionally examined by DTA and TG methods. Figure 2 shows DTA curve of the Fe0.83V1.17O4 phase, which contains two effects up to 1,000 °C. The first effect recorded on DTA curve is weak and exothermic, with onset temperature at 670 °C. This effect is accompanied by very weak, equal to 0.4 %, mass gain recorded on TG curve. The second effect registered on DTA curve is endothermic, with onset temperature at 850 °C.

Fragment of DTA curve (a) and TG curve (b) of Fe0.83V1.17O4 phase
In order to define the nature of the process connected with the exothermic effect recorded on DTA curve sample of Fe0.83V1.17O4 phase was heated for 1 h at 700 °C that is at temperature of maximum of the exothermic effect, and quenched to room temperature. Figure 3 shows fragments of powder diffraction patterns of phase Fe0.83V1.17O4 (curve a), phase Fe0.83V1.17O4 heated for 1 h at 700 °C (curve b), and pure phase FeVO4-I (curve c). The powder diffraction pattern of Fe0.83V1.17O4 sample after heat treatment at 700 °C for 1 h (Fig. 2, curve b) contains only the set of diffraction lines characteristic for FeVO4-I. It means that at 670 °C Fe0.83V1.17O4 phase undergoes a transition to phase of FeVO4-I structure. The distinctly pronounced endothermic effect registered on DTA curve of Fe0.83V1.17O4 with beginning temperature at 850 °C now can be undoubtedly attributed to the melting of phase of FeVO4-I structure. Small mass gain accompanying transition from Fe0.83V1.17O4 to the phase of FeVO4-I structure is probably connected with oxidation of V4+, V3+, or Fe2+ ions occupying some of the positions in the crystal lattice of Fe0.83V1.17O4. On this stage of investigations, one cannot exclude, however, the presence of oxygen vacancies in the crystal lattice of the phase under study. This problem will be the subject of further investigations.

Fragments of powder diffraction patterns of phase Fe0.83V1.17O4 (curve a), phase Fe0.83V1.17O4 heated for 1 h at 700 °C (curve b), and pure phase FeVO4-I (curve c)
The samples representing selected stages of Fe0.83V1.17O4 phase synthesis were subjected to an investigation with the use of infra-red spectroscopy (IR). Figure 4 shows IR spectra of initial mixture containing 75 mol% of V2O5 and 25 mol% of Fe2O3 (curve a), sample after heating at 900 °C for 3 h and quenched to room temperature (curve b), pure Fe0.83V1.17O4 phase (curve c), sample obtained after heating of Fe0.83V1.17O4 at 700 °C for 1 h (curve d), and pure FeVO4-I (curve e). IR spectrum of initial mixture containing 75 mol% of V2O5 and 25 mol% of Fe2O3 (curve a) reflects mainly IR absorption bands characteristic for V2O5. On the other hand, intensity of absorption bands characteristic for V2O5 in sample after heating at 900 °C for 3 h and quenched to room temperature (curve b) decrease considerably, what is accompanied with appearance of some new bands characteristic for Fe0.83V1.17O4 phase. IR spectrum of Fe0.83V1.17O4 phase given for the first time in this work (curve c) reveals the presence of absorption bands with their maxima at 946, 918, 826, 710, 648, 488, and 372 cm−1. Absorption bands covering wave number region of 1,000–550 cm−1 can be attributed to stretching vibrations of V–O bonds in VO4 tetrahedra [2, 21, 22]. Next absorption bands recorded in the range of 550–300 cm−1 can be the most likely ascribed to stretching vibrations of Fe–O bonds in MO6 octahedra and bending vibrations of M–O–M bridges [2, 21, 22]. Despite some similarities this spectrum differs from IR spectrum of FeVO4-I (curve e) what reflects difference in the crystal structure of both compounds. On the other hand, IR spectra of Fe0.83V1.17O4 phase heated at 700 °C for 1 h (curve d) and FeVO4-I (curve e) are very similar. As the both samples contain phases of FeVO4-I structure, it corroborates the results of X-ray qualitative phase analysis indicating that Fe0.83V1.17O4 phase transforms at 670 °C to phase of FeVO4-I structure.

IR spectra of initial mixture containing 75 mol% of V2O5 and 25 mol% of Fe2O3 (curve a), sample after heating at 900 °C for 3 h and quenched to room temperature (curve b), pure Fe0.83V1.17O4 phase (curve c), sample obtained after heating of Fe0.83V1.17O4 at 700 °C for 1 h (curve d), and pure FeVO4-I (curve e)
Conclusions
In this work, the results concerning the preparation and properties of a new Fe0.83V1.17O4 phase were presented for the first time. The XRD, DTA/TG, SEM/EDX, and IR measuring techniques were used to study properties of this phase. Fe0.83V1.17O4 is a dark brown solid which density equals to 3.94 g cm−3. It crystallizes in a monoclinic system and is isostructural with α-MnMoO4. At 670 °C Fe0.83V1.17O4 undergoes an irreversible transition to a phase of FeVO4-I structure. Despite some similarities, IR spectrum of Fe0.83V1.17O4 differs from IR spectrum of FeVO4-I what reflects differences in the crystal structure of both compounds.
References
Cheng F, Chen J. Transition metal vanadium oxides and vanadate materials for lithium batteries. J Mater Chem. 2011;21:9841–8.
Surca-Vuk A, Orel B, Drazic G, Colomban P. Vibrational spectroscopy and analytical electron microscopy studies of Fe–V–O and In–V–O thin films. Monatsh Chem. 2001;133:889–908.
Denis S, Baudrin E, Touboul M, Tarascon J-M. Synthesis and electrochemical properties of amorphous vanadates of general formula RVO4 (R = In, Cr, Fe, Al, Y) vs. Li. J Electrochem Soc. 1997;144(12):4099–109.
Ali EB, Bernede JC, Ray PL. Vanadium transition metal oxide films obtained by anneling under room atmosphere of metal layers sequentially deposited. J Cryst Growth. 2000;208:471–81.
Permer D, Laligant Y. Crystal structure of the tetrapolyvanadate Fe2V4O13. Eur J Solid State Inorg Chem. 1997;34:41–52.
Walczak J, Ziolkowski J, Kurzawa M, Osten-Sacken J, Lysio M. Studies on Fe2O3–V2O5 system. Pol J Chem. 1985;59:255–61.
Levinson LM, Wanklyn BM. Crystal growth and magnetic behaviour of FeVO4. J Solid State Chem. 1971;3:131–3.
Robertson B, Kostiner E. Crystal structure and Mössbauer effect investigation of FeVO4. J Solid State Chem. 1972;4:29–37.
Oka Y, Yao T, Yamamoto N, Ueda Y, Kawasaki S, Azuma M, Takano M. Hydrothermal synthesis, crystal structure, and magnetic properties of FeVO4-II. J Solid State Chem. 1996;123:54–9.
Laves F. On the high-pressure form of FeVO4. Acta Cryst. 1964;17:14761477.
Muller J, Joubert JC. Synthese sous haute pression d’oxygene d’une forme dense ordonnee de FeVO4 et mise en evidenced’une variete allotropique de structure CrVO4. J Solid State Chem. 1975;14:8–13.
Young AP, Schwartz CM. High pressure forms of CrVO4 and FeVO4. Acta Cryst. 1962;15:1305.
Attfield JP. The structure of the solid solutions formed in the system (Cr x Fe1−x )VO4. J Solid State Chem. 1987;67:58–63.
Attfield JP, Cheetham AK, Johnson DC, Novet T. Magnetic frustration spirals and short-range order in Cr x Fe1−x VO4-I solid solutions. J Mater Chem. 1991;1(5):867–73.
Touboul M, Denis S, Segui L. Crystal structure of a new form of chromium(III) vanadate(V), CrVO4-I. Eur J Solid State Inorg Chem. 1995;32:577–88.
Touboul M, Melghit K, Bernard P, Louer D. Crystal structure of a metastable form of indium orthovanadate, InVO4-I. J Solid State Chem. 1995;118:93–8.
Blonska-Tabero A. New phase in the system FeVO4–Cd4V2O9. J Therm Anal Calorim. 2008;93:707–10.
Blonska-Tabero A. Pb2Fe2V4O15—a new phase forming in the system FeVO4–Pb2V2O7. J Alloys Compd. 2010;508:42–6.
Bosacka M, Filipek E, Šulcova P, Dohnalová Ž, Paczesna A. Phase equilibria in the solid state and colour properties of the CuO–In2O3 system. J Therm Anal Calorim. 2012;109:605–10.
Blonska-Tabero A, Bosacka M, Dabrowska G, Filipek E, Piz M, Rychlowska-Himmel I, Tabero P, Tomaszewicz E. The synthesis and properties of the phases obtained by solid–solid reactions. J Min Metall. 2008;44B:19–26.
Dabrowska G, Tabero P, Kurzawa MJ. Phase relations in the Al2O3–V2O5–MoO3 system in the solid state, the crystal structure of AlVO4. J Phase Equilib Diffusion. 2009;30(3):220–9.
Tabero P. Formation and properties of the new Al8V10W16O85 and Fe8+x Al x V10W16O85 phases with M–Nb2O5 structure. J Therm Anal Calorim. 2010;101:561–6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.