
Abstract
Trace analysis (at fg-level) of radioisotopes requires a considerable push in analytical technology. Among most sensitive are a Time-Resolved Laser-Induced Fluorescence (TRLIF) and Chemiluminescence (TRLIC) methods for detection of elemental compositions and valence states and a Resonance Ionisation Spectroscopy (RIS) in combination with mass spectrometry for isotope composition determination. The radioisotopes of interest in environmental radiochemistry and planetary science and their analysis using TRLIF/TRLIC/RIS are discussed. The aspects of the development of the new technology implementing these methods are also described.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The use of laser radiation with tunable wavelength allows [1,2,3] selective excitation of actinide/lanthanide species with subsequent registration of luminescence or chemilumi-nescence for their detection. Using short laser pulses for excitation of molecules and ions in liquids and time resolution (TR) for registra-tion of luminescence (TRLIF) and chemiluminescence (TRLIC) produced by actinide and lanthanide ions one can efficiently separate target signals from short-lived background luminescence. Application of the multi-step scheme of luminescence/chemiluminescence excitation increase both the sensitivity and selectivity of the detection of substances [1,2,3]. The behavior of lanthanides/actinides in the environment determined by their molecular speciation, which can be detected by TRLIF/TRLIC. A combination of the TRLIF/TRLIC method with RIS laser and mass spectroscopy methods allows one to obtain more complete information about the sample.
Resonance ionisation spectroscopy (RIS) makes use of the laser light for resonance excitation of atoms into a higher energy state with its subsequent ionisation. The ionisation can proceed either into a continuum by use of e.g. powerful (> 1 mJ) pulsed laser beams, or electric field if the atoms are in the Rydberg states, or alternatively, the atoms can be excited into auto-ionising states located above the ionisation potential from where they self-ionise with high probability. The ionisation efficiency frequently can approach nearly 100% depending on the excitation scheme used. Combined with modern ion detection techniques e.g. mass spectrometry, the resonance ionisation provides a unique opportunity for study of the isotopes that are of a limited availability (few hundreds of atoms) similar to those produced in spallation reactions with protons at the isotope separators online e.g. at Isotope Resonance Ionisation Spectroscopy (IRIS) in Gatchina, Russia, or Isotope Separator Online Device (ISOLDE), in CERN, Geneva Switzerland [4]. The excitation schemes for the isotopes of many elements have been developed there and the work is still ongoing to cover the whole periodic table.
The trace analysis of isotopes with relative abundances of < 10−10 or those samples depleted in the isotopes of interests is another important application of RIS techniques. Conventional methods e.g. mass spectrometry, Inductively Coupled Plasma Mass Spectrometry (ICPMS), low-level counting (for radioactive isotopes) or X-Ray fluorescence spectroscopy (XRF) are not always sensitive for ultra-low concentrations and suffer from a lack of isobaric selectivity. In contrast, RIS coupled with e.g. time of flight mass spectrometry is capable of selective detection of < 1000 atoms of interest. Prof. J. Gilmour and Dr. I. Strashnov have developed such a setup for 81Kr at School of Earth Atmospheric and Environmental Science (SEAES) of Manchester University in 2010 [5].
This research is a result of collaboration across a range of scientific disciplines: nuclear and atomic physics, planetary science and cosmochemistry, earth and environmental science and radiochemistry where expertise in photon science is required for the development of the new state of the art techniques for detection of ultra-trace concentrations of isotopes [5, 6]. Several resonance ionisation facilities have been built around the world [7,8,9] (here we discuss a range of exciting new planetary and environmental radiochemistry research projects we have taken part in). These facilities combine resonance ionisation of atoms with pulsed and CW lasers (Nd:YAG, Ar-ion, tunable dye, and TiSa) with a time of flight mass spectrometer/RF qudrupole filter and utilize a state of the art technology available at the host institutions [10,11,12,13].
Environmental radiochemistry applications
According to the “The radioactive waste inventory” ~ 132 000 m3 of radioactive wastes have been accumulated in the United Kingdom (UK) to date [14]. These wastes are mainly a leftover after the spent fuel reprocessing cycle. It is expected their volume will significantly increase after implementation of new nuclear building programs. New policies currently being implemented in the UK, that includes the activities for identification of the potential sites for Geological Disposal Facilities of the nuclear wastes (GDF). Currently, all the wastes are placed in “the interim storage facilities”. They are supposed to be transported into a GDF in the future where they will be buried underground (200–1000 m) indefinitely. The identification of a suitable waste form (currently radioactive liquid wastes are vitrified to reduce their volume and prevent leakages), choice of buffer and backfill materials that will be placed around the waste packages in the excavation tunnels and the choice of suitable geological environment for GDF e.g. those with low groundwater flow containing the minerals that could sorb radionuclides from solution are among the main scientific goals. The research activities include the monitoring of existing storage facilities and the surrounding environment, and once the potential GDF sites are identified, construction of in situ rock testing laboratories will follow.
The radioactive wastes can be classified into the following categories. Firstly, the High-Level Wastes (HLW), that arises from PUREX process that involves the dissolution of spent nuclear fuel and other materials in nitric acid following chemical extraction of uranium and plutonium. These wastes are dominated by the heat-generating 90Sr and 137Cs. The Intermediate-Level Wastes (ILW), that do not generate significant amount of heat and contain the products from operation and decommission of nuclear power stations as well those left after spent fuel reprocessing e.g. aqueous wastes and organic materials used for U & Pu extraction, magnesium cladding, steel, zirconium alloy and graphite components of fuel. The radioisotopes of 237Np, 239Pu, 241Am, 99Tc, and 151Sm are dominant in these types of wastes. Low-Level Wastes (LLW) are the materials with radioactive content not exceeding 4 GBq/tonne of alpha or 12 GBq/tonne beta/gamma activity. They can include the items e.g. contaminated paper, plastic or metal products. Most of UK LLW are disposed of via shallow burial (near-surface) method. Typically 90Sr, 59Ni, 129I, and other isotopes can be detected in the LLW.
Development of the state of the art techniques capable of detection of trace amounts of radioisotopes in the environmental samples collected near nuclear power stations, fuel repossessing and decommission sites and wastes repositories is of a high priority for the environmental radiochemistry research [15]. In addition to environmental monitoring of these sites, detection of small concentrations of radioisotopes at the RIS facilities allows for studies of the migration behavior of these isotopes in different types of soil. For instance, understanding the role of colloids in radionuclide transport as well as of the seawater intrusion on their remobilization from the soil is of great importance. These studies, however, are difficult to perform in a typical radiochemistry laboratory involving the model experiments with only the most abundant minerals and high concentrations of radioactive isotopes. This poses a significant safety hazard. The RIS facilities, in contrast, require femtograms concentrations with activities not exceeding the natural background and thus are unique.
The isotope of interest and main objectives of laser ionisation mass spectrometry research:
-
90Sr: as a high yield product of fission of 235U and 238Pu it is the main tracer in the soil of the “problem” sites. It is chemically similar to Ca and once consumed incorporates strongly into the bones. Having pure beta-decay mode it is extremely difficult to detect with counting methods. Isobaric interferences with 90Zr and the presence of a stable 88Sr in excess of ~ 1010 make its measurements difficult with mass spectrometry. Resonance excitation with narrow-band continuous wave (CW) lasers can be employed, thus small concentrations of 90Sr will be selectively analysed.
-
134, 135, 137Cs: high yield products of 235U fission, all isotopes are readily absorbed in the soil. 137Cs can be used as a chronometer by measuring its daughter isotope (137Cs/137Ba ratio) e.g. in the studies of soil erosion and lake sedimentation. 135Cs/137Cs ratio is a characteristic of the reactor operating conditions because the production of 135Cs is sensitive to a neutron flux as its parent 135Xe is a good neutron absorber (σn = 2.6 × 106 b). In addition to radiochemistry research, the monitoring of 137Cs in foodstuff e.g. raw milk and meats and drinking water is recognized. Gamma spectroscopy is traditionally used for the detection of Cs, but high activities (or long measurement times) required due to sensitivities constrains.
-
99Tc: high yield fission product of 235U, highly mobile as able to form an anionic species, pertechnetate (Tc(VII)O4)− formed during spent fuel reprocessing is not easily extracted like Cs, U or Pu from the solution and the major concern in radioactive waste disposal due to solubility. Because of beta emission decay mode and isobaric interferences with Mo and Ru a low-level detection is difficult with conventional methods.
-
235, 236, 238U & 238−243Pu: their detection is of great importance as these two elements are the main components of the nuclear fuel. Their storage, especially of Pu and so-called “exotic” fuel from experimental and military reactors is challenging because of nuclear fission and proliferation issues. Separated in Sellafield in PUREX process Pu is kept in liquid form. In addition to environmental monitoring (Pu is highly toxic), the measurements of 235U/238U and 239Pu/240Pu ratios are important for nuclear nonproliferation.
Trace isotopes in planetary sciences
One of the fundamental questions the modern planetary science address—“How did our Solar system and its planets form and evolve?”—can be explored through the study of the abundances of the radioisotopes. At Manchester resonance ionisation facilities the extra-terrestrial samples are routinely analysed.
For instance, simulations show that some of the rocks ejected off Mars, Moon, and asteroids like Vesta by collisions with small bodies can gain the velocities high enough to escape their gravitational field and within several million years reach the Earth. This and the number of other observations are consistent with the identification of some meteorites as Martian. Because Mars lacks a magnetosphere the cosmic rays are not deflected, like on Earth, towards the poles inducing nuclear reactions in the regolith. The halogens (e.g. 37Cl/127I/79,81Br) and other elements (e.g. Rb, Sr, Zr) are converted into noble gases (38Ar/128Xe/80, 82Kr) by neutron capture, β-decay and spallation reactions. They can remain in the same mineral phase and due to their low chemical reactivity hold their history for millions of years. By analysing the isotope ratios of noble gases and other elements of different “bits” of the meteorites their formation and evolution history can be revealed.
Discoveries of isotopic anomalies in the meteorites have led to the identification of many inclusions (within these meteorites) e.g. chondrules and Ca–Al-inclusions (both are mm-size objects) that had been among the first materials solidified in the solar system. These anomalies imply the proto-solar nebula was not homogeneous and conflict with the models that assume a well-mixed cloud of elements with the same isotopic compositions. The microscopic (~ 1 μm diameter) SiC grains, that are highly enriched in s- and r-process isotopes are also present in the meteorites which point to a supernova explosion shortly before the formation of the solar system. The isotopic analysis (of Mg, Si, Ca, Ti, Sr, Ba, Nd, Sm, Te, Hg, U and extinct nuclides 244Pu, 107Pd, 26Al, and 22Na) of these objects are thus of a great interest albeit difficult due to their small size and trace concentrations.
A noble gas cosmochemistry is a modern branch of planetary science. Using technological advances in resonance ionisation mass spectrometry, several very high sensitivity mass spectrometers have been built in Manchester (RELAX and RIMSKI) [5]. They have been used for detection of several hundreds of atoms of noble gases in many applications:
The length of time a meteoroid has spent in space before arrival on Earth can be determined by comparing equilibrium abundance of radioactive 81Kr (t1/2 = 2.3 × 105 years), produced in nuclear spallation reactions with cosmic rays, and the concentration of stable cosmogenic Kr isotopes. The extreme sensitivity of RIMS allows for determining of these “cosmic ray exposure ages” of adjacent entities e.g. chondrules in meteorites. Exposure age variations between adjacent clasts must be due to exposure before incorporation into the host rock (i.e. in the early solar system). In this way, the irradiation environment of the early solar system can be studied.
Krypton and strontium record the environment of the s-process of nucleosynthesis. The relative abundances of Kr s-process isotopes depend on whether 79Se and 85Kr undergo neutron capture or beta decay (in the same way the relative abundance of 86, 87, 88Sr s-isotopes depends on its parent beta decaying 86, 87, 88Rb isotopes), which depend in turn on the temperature and neutron flux. Solar krypton and strontium appear to be a mixture of contributions from massive stars and Asymptotic Giant Branch (AGB) stars. Size-separates of presolar SiC and graphite grains show grain size dependent variation in the proportions of these components, but analysing multiple grains loses any information about individual stars. Analysis of Kr and Sr isotopes of individual presolar grains could allow for correlation with site-specific information determined from grain characterisation.
The meteorite named ALH 84001 is the oldest known Martian rock (~ 4 Ga). Mars lacks plate tectonics thus it has little modification. Because its noble gases composition is fractionated relative to that trapped in shock glass inclusions and Chassigny it has been argued it contains trapped paleoatmosphere [16]. The isotopic and elemental analysis of Xe and Kr of ALH 84001 is terra incognita. Comparison to the composition of its fluid inclusions will put more constraints on the models of the evolution of the Martian atmosphere.
The turnover of the lunar and Martian regolith by impacts from asteroids can be characterised using the different depths associated with implantation of the solar wind (millimetres), spallation production by cosmic rays (declining over ~ 1 m) and production by the capture of secondary neutrons (peaks at ~ 1 m). Kr and Sr isotopic studies of the regolith combined with the determination of the timing of exposure to secondary neutrons using 136Xe detection (it is produced in situ from fission of 232Th, 235U & 238U) will allow us to dramatically advance the understanding of lunar and Martian surface evolution.
Technical implementation: methodology and technology available
RIMS in planetary science
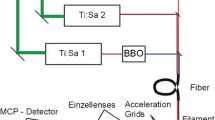
The laser ionisation mass spectrometer for the detection of Xe and Kr isotopes developed at The University of Manchester is regarded as one of the most sensitive in the world. Development of the RIMSKI (Resonance Ionization Spectrometer for Krypton Isotopes, Fig. 1) benefits from experience gained with RELAX (Refrigerator Enhanced Laser Analyser for Xenon) [17], which has been used for determination of isotopic ratios of Xe in various extraterrestrial samples [18]. Its ion source is a Wiley-McLaren design; the ions are detected by time of flight in a simple linear mode.

Schematic of the Resonance Ionisation Mass Spectrometer for ultra-trace detection of Krypton Isotopes (RIMSKI). The solids (meteorites/small grains) are step-heated by a 1064 nm CW Nd:YAG laser and the krypton atoms diffused out of the sample are introduced into the ion source of the time of flight mass spectrometer. The atoms then are condensed onto the cold substrate (70 K) placed behind the extraction electrode of the ion optics. The pulsed 1064 nm Nd:YAG laser beam evaporates the condensed atoms from the substrate, and the atoms are resonantly excited from the ground state into ionisation continuum by a set of pulsed ns dye lasers. The resonance ionisation scheme involves a vacuum ultraviolet 116.4 nm transition followed by 558.04 nm and 1064 nm
The schematic of the apparatus and the ionisation scheme in use is presented in Fig. 1. Small mg-size grains of meteorites can be laser heated in a vacuum (a 1064 nm CW Nd:YAG laser is used). Alternatively, samples of gas (e.g. laboratory air having a standard isotope composition) can be admitted into the mass spectrometer ion source after gettering for several minutes. The gas is condensed onto the cold stainless steel substrate at 70 K behind the extraction electrode from there it is evaporated by the laser pulse (1064 nm Nd:YAG, 10 ns). The background ions, notably hydrocarbons, can be produced as a result of the evaporation pulse. However, the noble gases, having higher ionisation potentials exist in the plume as neutrals.
After the delay of 600 ns, the Kr atoms are resonantly ionised by the three pulsed laser beams (116.48 nm, 558.04 nm, 1064 nm). The ionisation scheme was originally developed in Oak Ridge National Laboratory at Atom Sciences Inc. and the University of Tennessee [19]. The first transition takes place from the ground 4s24p5(2P°1/2)5 s, J = 1 state, and, as for all noble gases, lying in the vacuum ultraviolet region not accessible by the commercial lasers. A non-linear four-wave sum-frequency mixing in xenon-argon cell located between the ion source entrance and the laser system takes place. A powerful 1–2 mJ 252.5 nm beam excites the xenon gas in the cell via two-photon absorption and creates a dipole polarization in the gas. The argon is slowly added until the non-linear conditions created in the Xe–Ar mixture allowing for the sum-frequency mixing of the 252.5 nm and 1507.3 nm beams producing 116.48 nm photons [20]. The scheme, also technically complicated, is very selective and sensitive. The bandwidth of the dye lasers is comparable with the hyperfine structure of the krypton isotopes and much larger than their isotope shifts, hence all isotopes are excited and ionized simultaneously. Some isotope effects present and can be successfully addressed by choosing appropriate laser fluences and manipulations of the magnetic levels (with the λ/2 plate in the 558.04 nm beam) [21, 22].
The sensitivity of the apparatus of < 1000 krypton atoms is demonstrated in Fig. 2 where the typical time of flight mass spectra is presented. The laboratory air with known krypton composition is used for calibration. The mg-size grains from Sioux County and Pasamonte meteorites containing < 1000 81Kr atoms have been step-heated; krypton TOF spectra are presented in Fig. 2 b) and c) respectively. In both cases, a cosmogenic radioisotope 81Kr is detected (see enlarged TOF region) and can be used in the determination of the so-called cosmic ray exposure age of the meteorites or their time of arrival on Earth. Our other works further developed the applications of 81Kr isotopes and include detection of 81Kr-Kr exposure ages (relative measurements of 81Kr to stable Kr isotopes) of many meteorites which helped to identify the time of the impacts and their parent bodies [23]. The sensitivity was pushed even further and the age of the individual chondrules (that predate the formation of our solar system) were also identified [24, 25].

Resonance ionisation mass spectra of krypton isotopes from a laboratory air standards containing 106 krypton atoms, b Sioux County meteorite with a total weight of 3.3. mg containing roughly the same total amount of krypton atoms, c Pasamonte meteorite, which is a smaller grain of 1.2 mg with an order of magnitude less, 105 total krypton atoms
RIMS for detection of environmental radioisotopes
Another apparatus has been developed jointly by our Manchester and Moscow colleagues [26] and aimed at the ultra-trace analysis of uranium isotopes. It consists of a commercial time of flight Matrix-Assisted Laser Desorption Ionisation mass-spectrometer originally used for the detection of biomolecules using a Matrix-Assisted Laser Desorption Ionisation (MALDI) technique. The spectrometer’s ion source was modified and a laser system was introduced coupling the beams from pulsed dye-lasers. The solid samples (or dried on the graphite substrate liquids) can be desorbed with a pulsed nitrogen laser (3 ns pulse width, 337 nm, energy up to 300 μJ/pulse) focused into a ~ 20 µm diameter spot. When laser radiation interacts with the sample substrate, a plume of ions and neutral atoms is formed. Neutral atoms are resonantly excited and ionised by a laser photo-ionisation system. It is possible to automatically move the samples in the horizontal and vertical directions using stepper motors with ~ 22 µm steps. The photoionization laser system consists of two tunable dye lasers manufactured by Lambda Phtometrics and Sirah, pumped by the second (532 nm) and the third (355 nm) harmonics of a solid-state Nd:YAG laser. The Nd: YAG laser operates in the Q-switched mode. Part of the first harmonic (1064 nm) of the Nd: YAG laser is combined with the radiation from the two other lasers and also focused into the ion source. The ionisation process involves a resonance excitation from the ground state by 424.23 nm (~ 1 µJ/pulse) and subsequently by either 578.48 nm, 575.42 nm or 574.10 nm (~ 20 µJ/pulse) with 1064 nm (> 1 mJ) ionisation into a continuum. A three-color excitation, targeting specific uranium atomic levels, allows for selective ionisation of the uranium atoms. For instance, for the samples of depleted uranium the 235U/238U < 0.003 ratios was determined with < 7% precision (2σ errors) for the total uranium concentrations not exceeding ~ 80 fg/sample.
In the cases when the high selectivity is not required, the desorption ionisation signal can be used for the identification of uranium species, including the molecular compounds. Although the expected sensitivity is lower compared to the resonant ionisation mode with post-ionisation tunable dye lasers, the experiment is less complicated requiring just one 337 nm laser for both analyte evaporation and ionisation. This mode has been used in the studies of environmental samples. The clay mineral “kaolinite” (structural formula: Al2Si2O5(OH)4) is a soft white mineral. In the environment, there are granules of 0.1–10 micrometers. Due to its prevalence and a number of useful properties, such as high granularity, white color, and chemical inertness, it is widely used in industry as an ingredient in the manufacture of ink, organic plastics, rubber and cosmetics. Approximately 40% of the kaolinite currently mined is used in paper production. Due to its high melting point, kaolinite is also used in the manufacture of porcelain, ceramics and refractory materials. Currently, kaolinite is intensively mined in countries such as France, England, Germany, the Czech Republic and the United States [27]. It has been shown kaolinite readily absorbs uranium from the environment [28] playing an active role in isotopes migration processes. In the uranium RIMS experiments, kaolinite mined in the USA (KG0-1b) was used. Previously, the uranium concentration was determined by the inductively coupled plasma (ICP-MS) technique [29]. The content of 238U in the kaolinite studied was 1.6 ppm, and the 235U isotope was not detected. In our laboratory, the kaolinite granules were mixed with fullerene powder (12C60). Kaolinite reflects laser light at 337 nm and we found fullerene, depending on experimental conditions, enhance the photo-signal up to several orders of magnitude. After heating-shaking and ultrasonication steps the fullerene-kaolinites isopropanol suspensions were prepared and placed onto the synthetic graphite substrate. The ratio of fullerene and kaolinite in the prepared samples was 1: 2, and 20 ml of isopropanol accounted for 20 mg of the fullerene-kaolinite mixture in the preparation of suspensions. A typical time-of-flight spectrum of 1 ppm of uranium in kaolinite is presented in Fig. 3. Fullerene molecules (m/z = 720) are visible on the spectrum as well as their fragments (12C60-2n). The uranium atomic, as well as oxide species, are observed.

A desorption photoinisation time of flight mass spectrum of uranium in clay mineral kaolinite (1.6 ppm content) using non-resonant ionisation by 337 nm pulsed nitrogen laser. A fullerene 12C60 matrix was used for the enhancement of the photo-signal
TRLIF and TRLIC laser spectroscopy
After irradiation of the solutions with pulsed (10 ns) laser radiation, immediately after the laser pulse, a large luminescent background is observed with a characteristic luminescence time of 10−8 s. Since the characteristic luminescence lifetimes of the lanthanide and actinide series are 10−3–10−6 s, the signal can be recorded with a relatively large delay permitting for a significant reduction in the luminescence background using Time-Resolved Laser-Induced Fluorescence (TRLIF) methods.
Currently, the luminescence in solutions of actinides is not well known except UO2+, Am3+, Cm3+, Cf3+, Bk3+, and Es3+. The luminescence spectrum of lanthanides/actinides in solutions is usually recorded with a delay of 10−3–10−6 s with respect to the pump laser pulse (which is an optimal time window). The analysis requires about 1 ml of solution and the sensitivity of the method can reach up to 10–13 mol/l [1,2,3]. TRLIF has intrinsically high selectivity owing to four parameters i.e. the wavelength of the laser radiation, the wavelength of the detected radiation, the delay time and the duration of the luminescence recording time window.
TRLIF is well suited for analyzing inorganic samples. Biological samples and samples containing a significant amount of organic matter must be mineralized prior to analysis [3]. Our previous application of the TRLIF technique to biological samples (blood, urine) is described elsewhere [3]. Without mineralization, the limit of uranyl detection (LOD) in plasma using TRLIF was 0.1 ng/ml, and 8 pg/ml–10 pg/ml after mineralization. Mineralization did not affect the sensitivity of the detection of uranyl in the urine and we observed the detection limit of ~ 5 pg/ml. Since typical uranyl concentrations [3] in plasma are 0.05–0.5 ng/ml, and in urine 0.2–5 ng/ml, the detection limits reached by this method in our experiments allowed for robust determination of the presence of excess uranyl in the body. Circulation times of uranyl (and molecules of other elements) in the blood plasma and urine are different. Therefore, by measuring the dependences of uranyl concentrations on time in urine and blood plasma, it is possible to estimate the time of receipt of excess uranium (and other substances) in the body [1,2,3].
Luminescence of Pu, Np and some U compounds in liquids was not observed due to strong non-radiative relaxation. However, using chemiluminescence of solutions initiated by excited actinides/lanthanides, it is possible to develop detection techniques for Pu, Np, and U in liquids with high sensitivity, exciting actinides using a tunable pulsed laser [1,2,3].
The Limits of Detection (LOD) for spectrometers using chemiluminescence are in the range from 10−6 to 10−13 mol/l depending on the type of solutions and type of detectable molecule [3].
A lack of luminescence in solutions of Pu, Np, and some U compounds can be overcome with the use of laser excitation. In this case, these compounds can initiate chemiluminescence of chemiluminogen (luminol in our experiments) [1,2,3]. Two points are extremely important for the detection of actinides/lanthanides in solutions using chemiluminescent methods. The first is the presence of a delay of several microseconds between the maximum chemiluminescence intensity and the excitation laser pulse that makes it possible to apply Time-Resolved Laser-Induced spectroscopy methods (TRLIC) to detect actinides (e.g. U, Np, Pu). The second possibility is a use of multi-stage excitation schemes (in our experiments we used a two stage one) which warrants the high selectivity of a method.
Conclusions
RIMS is a powerful technique finding its applications in the detection of ultra-trace concentrations of various isotopes. Several laser ionisation facilities that are tackling problems ranging from the analysis of primordial noble gases extracted from meteorites, chondrules, and detection of radiogenic isotopes in the environment. The developed technology incorporates laser desorption/heating and cryogenic sample preconcentration, a range of tunable pulsed (ns or fs) lasers coupled into the ion sources of time of flight, magnetic sector or accelerator beamline facilities. The laser ionisation methods generally employ resonance excitation of the atomic levels of the targeted atoms/isotopes and are suitable for applications requiring high sensitivity and selectivity effectively eliminating background ion signals. A detection limit of < 100 atoms/sample has been demonstrated in routine analysis of radiogenic 81Kr from meteorites with precision and reproducibility of ~ 1% for the major isotopes.
The photoionisation processes are now well studied. Many efficient photoinisation schemes have been developed for various radionuclides e.g. 90Sr and Pu. For ionisation of strontium a double resonance excitation scheme from the ground state 5s2 1S0 → 5s5p 3P1 → 5s6s 3S1 with wavelengths of 689.45 nm and 688.02 nm followed by 488 nm beam from Ar-ion laser can be used [30]. Alternatively only the first transition followed by non-resonance absorption of the two photons at 488 nm can be used. The probability of the two-photon absorption from the first excitation step in Sr via a virtual level is enhanced because of 5s5d 3D1,2 levels located nearby. This scheme can be adopted when only one tunable laser is available. Pu can be efficiently ionised in the following scheme: 5d67s2 7F0 → 5f67s7p → 5f67s8s (586 nm and 688 nm from dye lasers, followed by 488 nm Ar-ion laser beam) [31].
Among very efficient methods of detection of actinides and lanthanides in solutions are those methods based on registration of actinides with time resolution, time-resolved laser-induced fluorescence (TRLIF) spectroscopy, having a limit of detection (LOD) up to 10−13 M.
Using laser radiation with tunable wavelength one can selectively excite various valence forms and molecules of actinides with subsequent registration of chemiluminescence (TRLIC). With knowledge of wavelength at which chemiluminescence appears and the intensity of chemiluminescence one can determine the concentration of a certain valence form of a given actinide and the structure of a complex containing this actinide. The LOD for spectrometers using the registration of chemiluminescence is in the range of 10−6–10−13 mol/l depending on the type of solutions and type of detectable molecule.
A combination of the TRLIF/TRLIC methods with other laser and mass spectroscopy methods allows one to obtain more complete information about the sample.
It is worth mentioning that a set of standard powerful techniques are also available. Electrospray/Atmospheric Pressure Chemical Ionisation coupled to electrostatic traps (Orbitraptm), Matrix-Assisted Laser Desorption Ionisation (MALDI), Gas/Liquid Chromatography and Inductively Coupled Plasma (Optical Emission/Mass-Spectrometry), the latter having the LOD down to ~ fg/ml, are all housed in our facility in Manchester.
References
Izosimov IN, Firsin NG, Gorshkov NG, Nekhoroshkov SN (2014) Detection of lanthanides and actinides in solutions based on laser-induced luminescence and chemiluminescence. Hyperfine Interact 227:271–281
Izosimov IN (2015) Application of multi-step excitation schemes for detection of actinides and lanthanides in solutions by laser spectroscopy. J Rad Nucl Chem 304:207–211
Izosimov IN (2016) Application of multi-step excitation schemes for detection of actinides and lanthanides in solutions by luminescence/chemiluminescence laser spectroscopy. Procedia Chem 21:473–480
Billowes J (2007) Proc.: conference on frontiers in nuclear structure, astrophysics, and reactions. Am Inst Phys: 213–219
Strashnov I et al (2011) A resonance ionization time of flight mass spectrometer with a cryogenic sample concentrator for isotopic analysis of krypton from extraterrestrial samples. J Anal At Spectrom 26(9):1763–1772
Wagia R et al (2018) Insight and control of the crystal growth of zeolitic imidazolate framework ZIF-67 by atomic force microscopy and mass spectrometry. Cryst Growth Des 18(2):695–700
Wagia R et al (2016) Determination of the preassembled nucleating units that are critical for the crystal growth of the metal-organic framework CdIF-4. Angew Chem Int Ed 55(31):9075–9079
Andrighetto A et al (2004) Proton-and neutron-induced fission on uranium carbide target. Eur Phys J A-Hadrons Nucl 19(3):341–345
Andrighetto A et al (2003) Production of neutron rich nuclides from uranium carbide targets of different density. Nucl Inst Methods B 204:267–271
Maul J et al (2004) A laser desorption/resonance enhanced photoionisation TOF-system for the spatially resolved trace analysis of elements. Nucl Instrum Methods Phys Res Sect B 226(4):644–650
Maul J et al (2006) Onset of crater formation during short pulse laser ablation. Appl Phys A 82(1):43–47
Maul J et al (2005) Multi-color resonance ionization of laser ablated gadolinium at high laser power. Opt Commun 256(4–6):364–372
Maul J et al (2006) Spatially resolved ultra-trace analysis of elements combining resonance ionization with a MALDI-TOF spectrometer. Anal Bioanal Chem 386(1):109–118
UK Radioactive waste & materials inventory: summary of data for international reporting. Department for Business, Energy and Industrial Strategy, Nuclear Decommissioning Authority. March 2017 ISBN:978-1-905985-35-7
NDA 5-year research and development plan 2019 to 2024. Nuclear Decommission Authority. 23 April 2019. Corporate report
Gilmour JD et al (1998) Xenon isotopes in irradiated ALH84001: evidence for shock-induced trapping of ancient Martian atmosphere. Geochem et cosmochem Acta 62(14):2555–2571
Gilmour JD et al (1991) A resonance ionisation mass spectrometer for xenon. Meas Sci Technol 2(7):589–595
Gilmour JD et al (2006) The I-Xe chronometer and early solar system. Meteorit Planet Sci 41(1):19–31
Thonnard N et al (1992) The second generation RIS_TOF noble gas detector: detection limits below 100 atoms in less than 5 minutes. Inst Phys Conf Ser 128:27–30
Strashnov I et al (2009) Tunable VUV light generation for resonance ionization mass spectrometry of Krypton. Opt Commun 282(5):966–969
Strashnov I et al (2009) Hyperfine structure induced isotopic effects in krypton resonance ionization mass spectrometry. Opt Commun 282(17):3487–3492
Strashnov I et al (2010) Controlling isotopic effects in the resonance ionisation mass spectrometry of krypton. Appl Phys B 99(3):543–551
Strashnov I et al (2013) Times of impacts that deliver samples of Vesta to Earth derived from ultrasensitive 81Kr–Kr cosmic ray exposure age analysis of Eucrites. Geochim Cosmochim Acta 106(1):71–83
Strashnov I et al (2013) 81Kr-Kr cosmic ray exposure ages of individual chondrules from Allegan. Meteorit Planet Sci 48(12):2430–2440
Strashnov I et al (2014) Resonance ionisation mass spectrometry of krypton and its applications in planetary science. Hyperfine Interact 227(1–3):259–270
Strashnov I et al (2019) A Laser Ablation Resonance Ionisation Mass Spectrometer (LA-RIMS) for detection of isotope ratios of uranium at ultra-trace concentrations from solid particles and solutions. J Anal At Spectrosc 34:1630–1638
Gaines R, Catherine H, Skinner W (1997) Dana’s new mineralogy: the system of mineralogy of James Dwifgt and Edward Salisbury, 8th edn. Wiley, Hoboken, pp 1–1407
Krepelova A, Sachs S, Bernhard G (2006) Uranium (VI) sotrption onto kaolinite in the presence and absence of humic acid. Radiochem Acta 94:825–833
Kogel JE, Lewis SA (2001) Baseline studies of the clay minerals society source clays: chemical analysis by inductively coupled plasma-mass spectroscopy (ICP-MS). Clays Clay Miner 49(5):387–392
Spectrochim Bushaw BA et al (1998) Diode-laser-based RIMS measurments of strintioum. AIP Conf Proc 454:177
Kunz P et al (2005) Resonance ionisation mass spectrometry (RIMS) with pulsed CW-lasers on Plutonium. Hyperfine Interact 162(1–4):159–166
Acknowledgements
This work was partially supported by the STFC GCRF fund [Grant Number ST/R002681/1].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Strashnov, I., Fernando, R. & Izosimov, I. Trace analysis of radioisotopes by laser spectroscopy and mass spectrometry. J Radioanal Nucl Chem 322, 1437–1445 (2019). https://doi.org/10.1007/s10967-019-06899-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06899-7