
Abstract
A new method of silica surface functionalization with carboxyl groups is proposed based on the reaction between 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid, MA) and the surface silanol groups. For the first time such a post-synthesis modification was successfully applied to silica materials. Although silica surface is successfully carboxylated (as evidenced by FTIR and XPS), the porosity, ordering and morphology remain practically unchanged after the proposed post-synthesis treatment (as confirmed by nitrogen sorption, XRD and electron microscopy). Adsorption study of model biomolecules (lysozyme and bovine serum albumin) shows that different surface chemistries (–OH vs. –COOH) lead to different affinities between the silica surface and sorbates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Functionalization of mesoporous silica materials with organic groups is crucial for majority of applications including adsorption [1,2,3,4], catalysis [5,6,7], drug delivery [8,9,10,11], sensing [12, 13]. There are two ways to achieve the desired surface functionalization via covalent bonding: (i) one-pot co-condensation and (ii) post-synthesis grafting.
The co-condensation method enables direct incorporation of functional groups into the silica mesoporous framework during the one-pot synthesis what is usually achieved by mixing functional monomer (or monomers) with the silica precursor so all of them are involved in the formation of silica framework. The co-condensation route has several advantages such as homogeneous distribution of the functional groups and precise control of their concentration [14]. However, a higher concentrations of functional groups usually result in deterioration of ordering and porosity. Moreover, different hydrolysis and condensation rates of both monomers can favour homo-condensation [15].
Another functionalisation strategy is post-synthesis grafting: a template-free material with exposed surface silanol groups is reacted with a modifying agent which is able to be grafted to that groups. This route leads to the hybrid materials containing desired functionalities on the silica surface. However, main disadvantages associated with this method are: (i) the limited control over the even distribution of the functional groups inside the pore walls and external particle surface, and (ii) the limited functionalization efficiency as it is dependent on the number of accessible functional groups on the silica surface.
So far many functional groups have been used to modify the silica surface by both one-pot co-condensation and post-synthesis grafting including amine [16,17,18,19], cyano [20, 21], thiol [2, 22,23,24], vinyl [25,26,27], phenyl [20, 26, 28], alkyl [29,30,31,32] and many others. Some studies have also focused on the functionalization with carboxylic groups [33,34,35,36,37,38]. The literature reports several methods of surface carboxylation, recently reviewed in the excellent paper [39]. The classical and most widely applied post-synthesis treatment to obtain carboxylated surface is the synthesis of cyanide-modified silica and subsequent hydrolysis of cyanide groups to carboxylic groups (using H2SO4 as a catalyst) The main drawback of this method is that the cyanoalkyltrialkoxysilane used as a second monomer (apart from TEOS) decreases the structural ordering of the final SBA-15 mesostructure [37, 40, 41]. Another frequently applied method of carboxylation silica is the use of co-condensation of carboxyethylsilanetriol sodium salt and tetraethoxysilane [36, 42]. Recently the reported method (not mentioned in the review [39]) is based on co-condensation of TEOS with of 5-(triethoxysilyl)-pentanoic acid [15].
Carboxylic groups are reactive ones easily interacting via hydrogen bonding which makes them good ligands for inorganic and organic species. The electrostatic interaction between –COOH groups and metal ions can enhance the interactions making the carboxy-functionalized silicas good sorbent and catalysts. Moreover, carboxylic groups accessible in the porous silica framework may also serve as anchoring sites for biomolecules (e.g. enzymes, proteins) broadening the range of their potential applications. The main drawback of carboxylated silicas is the high cost of functionalized organosilanes (e.g. cyanoethyltriethoxysilane, carboxyethylsilanetriol sodium salt). Herein a new simple method of silica surface functionalization with carboxyl groups is proposed based on the reaction between 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid, MA) and the surface silanol groups.
2 Experimental
2.1 Reagents
There were used the following compound: tetraethoxysilane (TEOS, 99%, Sigma-Aldrich), 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid, MA, 98%, Sigma-Aldrich), HCl (36%, POCH), ethanol (EtOH, 99.8%, POCH), toluene (99.5%, POCH), Pluronic P123 (P123, Sigma-Aldrich), lysozyme (LYS, Sigma-Aldrich), bovine serum albumin (BSA, Sigma-Aldrich). All chemicals were used as received, without further purification.
2.2 Synthesis of the ordered mesoporous silicas
Synthesis of the sample M0: 2 g of P123 was dissolved in 60 mL of 2 M HCl and 12 mL of deionized water with stirring at 40 °C. After 8 h of stirring TEOS (20 mmol) was added dropwise to the solution. The resulting mixture was stirred at 40 °C for 24 h and aged at 100 °C for next 24 h. The obtained white solid was washed and the P123 template was removed by refluxing the powder with 30% H2O2 (3 h, 50 mL per 1 g of the sample). Then the sample was thoroughly washed with deionized water (300 mL), and again dried overnight at 70 °C. H2O2 treatment was chosen as a method for the removal of the P123 template due to its simplicity, effectiveness and introduction of high number of surface silanol groups [43] which in the next step will be used for grafting of MA.
2.3 Modification of the sample M0 with Meldrum’s acid
0.15 g of the M0 sample was added to the 50 mL of toluene. After that 0.72 (0.005 M) of MA was added and the mixture was refluxed for 3 h at 110 °C in the nitrogen atmosphere. After that time the faintly yellowish solid was separated and soaked several times with CH3Cl and EtOH to assure that the unreacted species coming from the MA decomposition will be removed from the porous silica structure. Finally it was treated with boiling water for 3 h and dried overnight at 70 °C. The resulting sample was denoted M1.
2.4 Instrumental characterization
The CHN elemental analysis was performed using the Perkin Elmer CHN 2400 analyzer. X-ray diffraction (XRD) patterns were recorded using the Empyrean diffractometer (PANalytical) (with 0.01° and 20 s steps in the range of 0.6° < 2θ < 6.0°). SEM and TEM analyses of randomly selected parts of the surface were performed by means of the LEO SEM 1430 VP and Carl Zeiss EM microscopes, respectively. The nitrogen sorption isotherms were measured at − 196 °C using Quantachrome Nova 1200e analyser. The samples were degassed in vacuum at 120 °C for 8 h prior to the analysis. The BET specific surface areas (SBET) were evaluated in the range of relative pressures of 0.05–0.20. The total pore volumes (Vt) were calculated by conversion of the amount adsorbed at relative pressure ~ 0.99 to the volume of liquid adsorbate. The average pore sizes (dBJH) and pore sizes distributions were estimated using the BJH [44] and KJS [45] methods using adsorption branch of the isotherm. FTIR spectra were recorded by means of FTIR 6200 spectrophotometer (Jasco) in the range of 4000–400 cm−1 with the resolution 4 cm−1 (32 scans were averaged for each spectrum). XPS spectra were obtained by using Multi-Chamber Analytical System (Prevac) with a monochromatic 450 W Al K-alpha X-ray radiation source (1486.6 eV) (Gammadata Scienta). The surface pH of the samples was measured using TitroLine alpha + titrator: 0.2 g of the sample was mixed with 30 mL of water and stabilized overnight before measurement.
2.5 Adsorption measurements
In every batch experiment 10 mg of adsorbent was shaken with 30 mL of buffered (25 mM acetate, phosphate or dicarbonate) solution of LYS or BSA at room temperature for 24 h. After the adsorption equilibrium had been achieved, the solution was separated from the sorbent by filtration using syringe 0.45 µm filter. The equilibrium adsorption amounts were calculated using a simple mass balance: a = (ci − cj) V m−1, where ci is the initial concentration of LYS/BSA (mg L−1), cj is the equilibrium concentration (mg L−1), V is the volume of the solution (30 mL) and m is the mass of the adsorbent (g). Measurements of LYS/BSA concentrations were made using the UV–Vis spectrometer SPECORD 200 (Analytic Jena) at the wavelength 283 nm (LYS) and 281 nm (BSA).
3 Results and discussion
3.1 Meldrum’s acid as a functionalization agent
2,2-Dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid, abbreviated MA throughout the text) is a cyclic diester with strongly acidic properties. It has been extensively used in the synthesis of both a synthetic building block and a thermolytic precursor to dialkyl ketenes [46,47,48]. The former was reported before as a tool for carboxylation of carbon fibers [49, 50]. Two different mechanisms presented in Fig. 1 were proposed. The first one is based on a nucleophilic attack of the carbonyl carbon atom (positions 4 and 6) by the surface hydroxyl group which induces a ring opening reaction to form the malonic acid ester followed by a proton transfer [49]. The second one is based on the reaction of surface carbonyl group carbon atom (position 1) of Meldrum’s acid to generate an intermediate with the carbon–carbon double bond through dehydration followed by ring opening and formation of carboxylic ester and acetone [50]. Considering the first scenario it would be possible to apply it to silica surfaces where significant amounts of hydroxyl groups are present on the surface and could possibly act as nucleophile agents. The polar silica surface with an abundant amount of silanol groups acting as anchoring points for Meldrum acid molecules could finally lead to its carboxylation. As the surface of silica has no carbonyl groups, it is hypothesized that the reactions described by the first mechanism could only occur in the case of pure silica sample. Although the synthesis was conducted at elevated temperature (reflux at 110 °C—the boiling point of toluene) most probably this temperature was not high enough to provide the formation of ketenes. It was reported that T > 200 °C is needed a thermal decomposition of the Meldrum’s acid ring yielding the dialkyl ketene along with the loss of acetone and carbon dioxide [47, 48]. Interestingly the color of the synthesis mixture (MA dissolved in toluene) was continuously changing during the 3 h treatment from the colorless solution via yellowish orange till the brown one. The origin of this phenomenon has not been explained in the literature yet.

Two reported routes of MA attachment to the surface of carbon fibers via hydroxyl (a) and carbonyl (b) groups
3.2 Chemical changes upon Meldrum acid treatment
Both samples, initial and carboxylated, were characterized by a broad range of instrumental techniques to get a detailed view of their ordering (XRD, TEM), porous structure (nitrogen sorption, TEM), chemical composition (elemental analysis, XPS, FTIR, surface pH) and morphology (SEM, TEM).
As it can be seen in Table 1 the initial sample M0 is composed of only silica and oxygen (hydrogen can not be detected by XPS) while in the case of modified sample M1 the content of carbon is 2.3%. Deconvolution of C 1s energy core level (Fig. 2) shows contributions of various C-species, among them, there is a signal corresponding to the O=C–O– bond at 289.3 eV clearly testifying to the presence of carboxyl groups on the silica surface. This is also supported by the presence of the signals characteristic of the C–O, C=O and C=C species. The presence of the XPS C 1s energy core level at 284.0 eV, characteristic of the sp2 carbon bonding, most probably comes from the adventitious toluene residues that were not able to be removed from the silica matrix.

Deconvolution of C 1s XPS signal of the sample M1
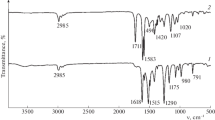
The bulk elemental analysis presented in Table 1 also shows higher carbon contents of both samples than XPS. This is due to the fact that XPS shows only surface composition of several nanometers while the elemental analysis gives the bulk composition. Apparently, both silicas contain unremoved traces of polymeric template trapped in the micropores hardly accessible to the oxidative agent (H2O2). Those pores are located in the bulk of the materials so XPS—opposite to bulk elemental analysis—cannot detect the trapped P123. This finding is confirmed by FTIR spectroscopy (Fig. 3) which is a very sensitive method for detecting light elements. Thus it can be expected that even a low content of carbon will be manifested on the spectra of final materials. Indeed, the presence of the –СН2– and –CH3 units of the template is confirmed by a group of bands in the region of 2830–2980 cm−1 (stretching modes of CH2 and CH3 groups) as well as low intensity band at 1460 cm−1 (deformation modes of CH2 and CH3 groups). Interestingly, after the MA treatment, signals located in the region 2830–2980 cm−1 are less intensive which indicates that a portion of residual P123 was removed during the applied high temperature (110 °C) treatment in toluene. Obviously, a small amount of toluene is strongly adsorbed in the micropores after the treatment with MA. This is confirmed by the signal at 670 cm−1 appearing after the functionalisation. This signal can be attributed to the aromatic C–H “out-of-plane” bending. This is supported by the presence of the sp2 species in C 1s XPS signal coming from adventitious toluene adsorbed in the micropores. Mirji et al. showed that toluene can be strongly adsorbed by SBA-15 silica and the temperature as high as 470 °C is needed to remove completely toluene from the porous structure [51]. As hypothesized by the authors toluene is rather decomposed not desorbed which testifies that it can be strongly adsorbed in the silica porous structure. Despite the removal of a portion of Pluronic the volume of micropores of M1 is 10% smaller than M0 (0.37 vs. 0.41 cm3, cf. Table 1) supports the hypothesis that toluene can be strongly bonded in the micropores what results with significant 10% loss of microporosity of M1 sample.

FTIR spectra of the samples M0 and M1
A wide absorption band in the region of ~ 1000–1150 cm−1 with a shoulder at ~ 1150–1300 cm−1 is unambiguously attributed to the stretching modes of the siloxane framework. This is supported by the presence of the bands at ~ 960 and ~ 800 cm−1 assigned to the out-of-plane bending modes of OH as well as O–Si–OH fragments, respectively [14].
Interestingly, after modification a low intensity band at 3735 cm−1 appears. This band comes from the stretching modes of silanol groups, ν(Si–OH). Obviously, the high temperature treatment causes breakup of hydrogen-bonded silanols. Adsorbed water is removed from silica surface leaving some amount of free silanol groups.
After modification with MA a new signal appears at 1720 cm−1, which can be undoubtedly attributed to the carbonyl stretching modes ν(C=O). In the FTIR spectrum of MA two bands at 1795 and 1755 cm−1 are found which is a characteristic feature of anhydrides. In the case of the sample M1 there is no such bands which is a clear indication of transformation of Meldrum acid to intermediate species and their subsequent grafting on the silica surface.
Surface pH of both samples is different—in the case of M0 sample the surface pH is 5.01 while for M1—4.44. That indicates that more acidic species are on the surface of carboxylated M1 sample dissociating to anions and protons leading to pH value lowering. Taking into account the composition of the synthesis mixture, the only one type of possible functional group are carboxylic ones. Thus the successful Meldrum’s acid modification was confirmed not only by XPS and FTIR spectroscopy but also by measurement of surface pH.
It should be clearly emphasized here that the goal of this research was to show possibility of applying Meldrum’s acid as a modification agent but not to optimize fully the proposed treatment scheme. It seems obvious that by varying reaction conditions such as time of the treatment, temperature, type of solvent, amount of MA, etc. it would be possible to tailor the functionalization efficiency (i.e. the amount of carboxyl groups attached to the silica surface).
3.3 Structural and morphological changes upon the Meldrum’s acid treatment
The low-angle XRD patterns for both samples (Fig. 4) exhibit three well-resolved diffraction peaks at 2θ = 0.86°, 1.44°, and 1.67° (for M0) and 2θ = 0.82°, 1.41°, and 1.64° (for M1). They are indexed as (100), (110), and (200), respectively, and are characteristic of hexagonal symmetry p6m. Only a slight decrease of the reflections intensities is being observed after functionalization. Obviously, the hexagonal ordered structure is well retained well after the MA treatment, indicating that the introduction of carboxyl groups does not disturb the mesoscopic ordering of a hexagonal structure.

XRD diffractograms of the samples M0 and M1
The nitrogen adsorption–desorption isotherms of the samples are shown in Fig. 5a and can be classified as IV type according to the IUPAC classification [52]. The isotherms of both samples exhibit hysteresis loops at p/p0 ≈ 0.65–0.8 due to the presence of uniform mesopores. The hysteresis loop of the sample M1 is slightly shifted towards lower values of relative pressure which indicates that the mesopore sizes are smaller. Indeed, the BJH average pore sizes are 10.4 and 9.8 nm for M0 and M1, respectively. The smaller pore size of M1 is due to the attachment of MA fragments. Interestingly, the pores sizes calculated by empirical KJS method which is recommended for ordered mesoporous silicas are similar to those obtained by BJH one (cf. Table 1). The specific surface area (SBET), total pore volume and micropore volume are also slightly smaller after functionalization (cf. Table 1). BJH pore size distributions (PSD) curves are presented in Fig. 5b showing the narrow distributions of pore sizes. Again, BJH and KJS method derived PSD curves are similar.

Nitrogen sorption/desorption isotherms of the samples M0 and M1 and corresponding pore size distributions
The SEM images presented in Fig. 6 show that both silicas, pristine and carboxylated, have typical morphology: hexagonal fiber-like cylinders arranged into bigger longitudinal structures composed of parallelly oriented cylinders. The analysis of TEM images presented in Fig. 7 reveals that each cylinder is formed by the ordered mesoporous network of parallelly oriented uniform mesopores with the diameter of several nm. That is consistent with the pore sizes obtained from the nitrogen sorption data (cf. Table 1). The morphology and microscopic structure of both samples are very similar which is also supported by XRD and nitrogen sorption data. Thus the proposed carboxylation scheme does not affect the morphological and structural properties of SBA-15 silica.

SEM images of the samples M0 and M1

TEM images of the samples M0 and M1
3.4 Sorption of LYS and BSA proteins
To investigate a possible effect of surface carboxylation on sorption performance of the resulting material, sorption of two proteins: lysozyme (LYS) and bovine serum albumin (BSA) was studied. Sorption of proteins is known to be very sensitive to changes in electrostatic interactions which in that case are tuned by modification surface chemistry (i.e. carboxylation). The probe proteins were chosen for their different size and physical properties which are summarized in Table 2 [53, 54]. As it can be seen both biomolecules have different sizes but they are smaller than the pores diameters of M0 and M1 silicas. Some steric restrictions inhibiting proteins access to the primary mesopores can be however more pronounced in the case of larger BSA molecule since sorption of one or two molecules at the pore entrance can block that pore for other molecules. Hover at lower concentrations of proteins this effect does not affect seriously the sorption uptakes due to big number of accessible adsorption sites. The pore diameters as well as other parameters of the porous structure are very similar for both silicas (cf. Table 1), as well as the experimental conditions of sorption process (ionic strength, etc.). Thus it can be anticipated that any differences in sorption behaviour can be attributed mainly to the role of the specific surface chemistry of both samples.
Adsorption of proteins onto mesoporous materials was reported to be governed by a wide range of surface–protein noncovalent interactions including electrostatic interactions, hydrogen bonding, hydrophobic and π–π overlapping [55]. In general, all the above-mentioned interactions can be controlled by specific surface modification. In the case of modification described in this paper, carboxylic groups can interact with amine groups of the proteins via the Coulombian electrostatic interactions as well as hydrogen bonding formation.
The effect of pH on adsorption uptake is shown in Fig. 8. According to the obtained results the maximum adsorption uptakes is observed at pH ≈ 5 for BSA and pH ≈ 11 for LYS. The maximum pH value for LYS adsorption can be beyond the range of investigated pH values as silica starts to dissolve at alkaline pH (there is a study reporting that even at pH > 7 the silica matrix becomes very unstable due to dissolution [56]). Despite this fact it can be assumed that the optimal pH values are close to the isoelectric points, pI, of both proteins (cf. Table 2) indicating the presence of electrostatic interactions between the protonated amino groups of the proteins and the hydroxyl and/or carboxyl groups on the silica surface. The additional reason for pH-dependency can be conformational changes (folding and unfolding of the proteins). Both explanations have been very comprehensively discussed by Azevedo and co-workers [57].

Influence of pH on adsorption of BSA and LYS
It is also worth noting that in the case of BSA dependences of adsorption uptake on pH the same trend is observed for both samples, while in the case of LYS there is a more pronounced difference in the range of pH = 7–9. The reason for that phenomenon can be restricted accessibility of bulky BSA molecules to the carboxyl groups located mainly in the primary mesopores: BSA molecule “feel” the surface chemistry of both silicas as very similar (no differences in uptake = f(pH) dependences, cf. Fig. 8). Smaller LYS molecules can enter more deeply into the mesopores and their interactions with carboxyl groups are more pronounced which results in different curves for uptake = f(pH) for M0 and M1.
Figure 9 shows the adsorption isotherms of LYS and BSA obtained in the batch mode at pH = 7. Such pH value was chosen as it is close to the physiological pH (7.365), thus it can reflect the sorption behaviour occurring under the conditions closer to real ones. An abrupt increase of the adsorbed amount is observed at low initial concentrations (ce < 1 g L−1) indicating a high affinity between the silica surface and proteins. At higher equilibrium concentrations (ce > 1 g L−1) all isotherms reach a plateau and adsorption uptake does not increase any more.

Adsorption isotherms of LYS (a) and BSA (b) on the studied samples measured at pH = 7 and their fitting to Langmuir and Freundlich models
The obtained sorption data were fitted with two adsorption models: Langmuir (Eq. 1) and Freundlich (Eq. 2):
where qm (mg g−1) is the maximum adsorption capacity, c (mg L−1) is the equilibrium solute concentration, and KL, and KF are the Langmuir and Freundlich constants, respectively. The parameter n is the factor characterizing the quasi-Gaussian energetic heterogeneity of the adsorption system. KL and c0 are the Langmuir constant and the highest initial concentration of metal ion used in this study, respectively. The calculated parameters are given in Table 3. Observed correlation coefficients showed that Langmuir model fit the results better than Freundlich model. This indicates that adsorption of both proteins onto pristine and carboxylated sample is a single-molecular-layer adsorption process. In Eq. 1 KL is the Langmuir constant related to the adsorption energy and its important feature is that it can be used to predict the affinity between the sorbate and sorbent using a dimensionless constant called the separation factor (RL). Its values calculated using Eq. 3 are also presented in Table 3. In general, if RL > 1, the adsorption is unfavorable, RL = 1 corresponds to the linear adsorption isotherm, while RL < < 1—the adsorption is favorable. If RL ≅ 0—adsorption is irreversible [58, 59]. Observed RL values for all studied samples are in the range of very small indicating a very favorable adsorption process. In the case of the sample M1 there are more than two times lower than those for the sample M1 indicating that the affinity of both proteins is higher when the surface is carboxylated.
This high affinity is reflected in the fact that any significant desorption of proteins in water was observed. When copious volumes of 0.9% NaCl solution were used a small fraction (< 5%) of the protein was desorbed. Certainly, in the case of hydrophilic surfaces, the ionic strange and pH must play some role in the desorption process, however surface denaturation can play a crucial role in that poor reversibility [60]. As it was nicely summarized there: “the concepts of reversibility and reconformation are closely related in protein adsorption, where the surface acts analogous to a heterogeneous catalyst to facilitate dramatic structural alteration of the protein resulting in an energetically favorable state” [60].
Comparing the static sorption capacities (SSC) given in Table 3 it can be seen that (i) sorption capacities for the sample M1 (255 mg g−1 for LYS, 42 mg g−1 for BSA) are slightly higher than for M0 (240 mg g−1 for LYS, 31 mg g−1 for BSA), (ii) SSC values are well predicted by Langmuir model (Table 3: compare calculated Qmax values vs. experimental SSC values), (iii) SSC values are similar to those observed in the literature at similar conditions [53, 55, 61,62,63].
Uptakes of LYS are considerably higher than those of BSA, mainly due to sterical restrictions for BSA. The Stokes hydrodynamic diameter of BSA is 8 nm in the range of pH 4–9 [64] so BSA is theoretically able to enter the mesopores. However, it is possibly stacked at the pore entrance blocking the passage of other molecules; thus it is adsorbed mainly on the external surface area [65]. In contrast, smaller LYS molecules can be adsorbed in the mesopores leaving enough space for other LYS molecules to enter the pore and be adsorbed in the interior.
The adsorption uptakes measured at different pH values from two concentrations (0.25 and 2.5 g L−1) are presented in Fig. 10. The first one provides a relatively low protein concentration which makes the protein–surface interactions predominant over the protein–protein ones. The second concentration is high enough to consider the adsorbed amount as the saturation value (plateau on the isotherms). As it can be seen from the Fig. 10 the uptakes from both concentrations are usually higher for the carboxylated sample M1. The differences between both samples are particularly noticeable for LYS at pH 7 and 9 (Fig. 10a) and BSA at pH 5 and 7. The adsorption of BSA from 0.25 g L−1 solution at pH = 7 is larger by 65% due to the altered interactions between the carboxylated surface and the protein.

Uptakes of LYS (a) and BSA (b) at different pH values from two initial concentrations: 0.25 and 2.5 g L−1
The question arises why at pH = 7 adsorption of BSA (negatively charged at pH > 5) is not prevented despite unfavourable electrostatic interactions (both BSA and silica are negatively charged at pH = 7). Obviously electrostatic interactions alone can not govern exclusively the adsorption behaviour and the specific interactions (e.g. formation of hydrogen bonds) and hydrophobicity should be also taken into account. More than 20 years ago it was suggested that the solvent-induced interactions (formation of hydrogen bonds, possibly involving water molecules mutually bonded by the protein and the silica surface) can be a main driving force for adsorption of globular proteins onto the hydrophilic silica surface [66]. The carboxylated surface of M1 sample contributes to the formation of hydrogen bonds (also water-mediated as predicted by molecular simulations [64, 66]). On the other hand, it was found that hydrogen bonding itself is not sufficient to drive LYS adsorption [55]. The observed adsorption of BSA onto the M0 sample at unfavourable pH = 7 can be also explained by the presence of positively charged fragments on the BSA surface which can enhance adsorption despite total negative charge of the molecule [63, 67]. Thus combination of specific and nonspecific interactions as well as other factors is responsible for the adsorption of protein onto silica surfaces, particularly functionalized ones.
4 Conclusions
A new post-synthesis method of carboxylation of mesoporous silica was proposed based on the reaction between 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid) and surface silanol groups. The resulting SBA-15 porous structure, ordering and morphology is almost intact as evidenced by nitrogen sorption, XRD, SEM and TEM measurements. In contrast, surface chemistry is successfully altered and the presence of carboxyl groups was confirmed by XPS and FTIR spectroscopy. The new method is based on the reaction of silica surface with purely organic reagent instead of functionalized silica monomer.
The adsorption of model biomolecules (lysozyme and bovine serum albumin) is affected by the designed surface chemistry. Saturation uptakes of pristine and functionalized SBA-15 silicas are rather similar for LYS (240 vs. 255 mg g−1) differ for BSA (31 vs. 42 mg g−1). However, the differences are more pronounced at lower concentrations for both LYS (135 vs. 176 mg g−1) and BSA (22 vs. 36 mg g−1), indicating that the presence of carboxylic groups affects the adsorption process. This can be due to not only enhanced electrostatic interactions but also hydrogen bonding between the surface groups and amine groups of proteins.
The proposed method can be exploited for fabrication of carboxylated silica surfaces as well as tuning the surface–protein interfacial interactions via tailoring functionalization of the surface.
References
T.X. Bui, S.-Y. Kang, S.-H. Lee, H. Choi, J. Hazard. Mater. 193, 156 (2011)
M. Barczak, J. Dobrzyńska, M. Oszust, E. Skwarek, J. Ostrowski, E. Zieba, P. Borowski, R. Dobrowolski, Mater. Chem. Phys. 181, 126 (2016)
M. Barczak, M. Wierzbicka, P. Borowski, Microporous Mesoporous Mater. 264, 254 (2018)
P.N.E. Diagboya, E.D. Dikio, Microporous Mesoporous Mater. 266, 252 (2018)
D. Farrusseng, A. Tuel, New J. Chem. 40, 3933 (2016)
P. Bhanja, A. Bhaumik, Fuel 185, 432 (2016)
S. Singh, R. Kumar, H.D. Setiabudi, S. Nanda, D.-V.N. Vo, Appl. Catal. A 559, 57 (2018)
M. Vallet-Regí, M. Colilla, I. Izquierdo-Barba, M. Manzano, Molecules 23, 47 (2017)
M. Manzano, M. Vallet-Regí, J. Mater. Sci. Mater. Med. 29, 65 (2018)
E. Juère, F. Kleitz, Microporous Mesoporous Mater. 270, 109 (2018)
J.G. Croissant, Y. Fatieiev, A. Almalik, N.M. Khashab, Adv. Healthc. Mater. 7, 1700831 (2018)
E. Rossinyol, J. Arbiol, F. Peiró, A. Cornet, B. Tian, T. Bo, D. Zhao, Sens. Actuators B 109, 57 (2005)
V.K. Tomer, P.V. Adhyapak, S. Duhan, I.S. Mulla, Microporous Mesoporous Mater. 197, 140 (2014)
M. Barczak, J. Solid State Chem. 258, 232 (2018)
A. Feinle, F. Leichtfried, S. Straßer, N. Hüsing, J. Sol-Gel. Sci. Technol. 81, 138 (2017)
Y. Zhu, H. Li, Q. Zheng, J. Xu, X. Li, Langmuir 28, 7843 (2012)
J. Aguado, J.M. Arsuaga, A. Arencibia, M. Lindo, V. Gascón, J. Hazard. Mater. 163, 213 (2009)
R. Dobrowolski, M. Oszust-Cieniuch, J. Dobrzyńska, M. Barczak, Colloids Surf. A 435, 63 (2013)
M. Barczak, R. Dobrowolski, J. Dobrzyńska, E. Ziȩba, A. Da̧browski, Adsorption 19, 733 (2013)
M. Barczak, E. Skwarek, W. Janusz, A. Dabrowski, S. Pikus, Appl. Surf. Sci. 256, 5370 (2010)
M. Oszust, K. Gdula, P. Borowski, A. Dąbrowski, M. Barczak, Adsorpt. Sci. Technol. 33, 669 (2015)
A. Bibby, L. Mercier, Chem. Mater. 14, 1591 (2002)
A. Arencibia, J. Aguado, J.M. Arsuaga, Appl. Surf. Sci. 256, 5453 (2010)
T. Kang, A. Park, Y, and Y. Jongheop, Ind. Eng. Chem. Res. 43, 1478 (2004)
Q. Wei, H.-Q. Chen, Z.-R. Nie, Y.-L. Hao, Y.-L. Wang, Q.-Y. Li, J.-X. Zou, Mater. Lett. 61, 1469 (2007)
M. Barczak, A. Da̧browski, S. Pikus, J. Ryczkowski, P. Borowski, M. Kozak, Adsorption 16, 457 (2010)
M. Barczak, S. Pikus, B. Skrzydło-Radomańska, A. Da̧browski, Adsorption 15, 278 (2009)
H.H.P. Yiu, P.A. Wright, N.P. Botting, J. Mol. Catal. Enzyme 15, 81 (2001)
J.C. Doadrio, E.M.B. Sousa, I. Izquierdo-Barba, A.L. Doadrio, J. Perez-Pariente, M. Vallet-Regí, A. Yli-Urpo, J. Kiesvaara, M. Marvola, J. Mater. Chem. 16, 462 (2006)
T. Yasmin, K. Müller, J. Chromatogr. A 1218, 6464 (2011)
M. Barczak, A. Dąbrowski, M. Iwan, Z. Rzączyńska, J. Phys. Conf. Ser. 146, 12002 (2009)
C.-M. Yang, H.-A. Lin, B. Zibrowius, B. Spliethoff, F. Schüth, S.-C. Liou, M.-W. Chu, C.-H. Chen, Chem. Mater. 19, 3205 (2007)
Z. Bahrami, A. Badiei, G.M. Ziarani, J. Nanoparticle Res. (2015). https://doi.org/10.1007/s11051-015-2929-1
P. Valle-Vigón, M. Sevilla, A.B. Fuertes, Microporous Mesoporous Mater. 176, 78 (2013)
Q. Yang, S. Wang, P. Fan, L. Wang, Y. Di, A.K. Lin, F.-S. Xiao, Chem. Mater. 17, 5999 (2005)
C.H. Tsai, W.C. Chang, D. Saikia, C.E. Wu, H.M. Kao, J. Hazard. Mater. 309, 236 (2016)
Y.C. Pan, H.Y. Wu, L.P. Lee, G.L. Jheng, G.T.K. Fey, H.M. Kao, Microporous Mesoporous Mater. 123, 78 (2009)
W. Bian, L.L. Lou, B. Yan, C. Zhang, S. Wu, S. Liu, Microporous Mesoporous Mater. 143, 341 (2011)
L. Han, O. Terasaki, S. Che, J. Mater. Chem. 21, 11033 (2011)
S. Shen, P.S. Chow, S. Kim, K. Zhu, R.B.H. Tan, J. Colloid Interface Sci. 321, 365 (2008)
C. Yang, Y. Wang, B. Zibrowius, F. Schüth, Phys. Chem. Chem. Phys. 6, 2461 (2004)
L. Han, Y. Sakamoto, O. Terasaki, Y. Li, S. Che, J. Mater. Chem. 17, 1216 (2007)
M. Barczak, N. J. Chem. 42, 4182 (2018)
E.P. Barrett, L.G. Joyner, P.P. Halenda, J. Am. Chem. Soc. 73, 373 (1951)
M. Jaroniec, L.A. Solovyov, Langmuir 22, 6757 (2006)
F.A. Leibfarth, M. Kang, M. Ham, J. Kim, L.M. Campos, N. Gupta, B. Moon, C.J. Hawker, Nat. Chem. 2, 207 (2010)
F.A. Leibfarth, M. Wolffs, L.M. Campos, K. Delany, N. Treat, M.J. Kade, B. Moon, C.J. Hawker, Chem. Sci. 3, 766 (2012)
J. Wu, S.T. Iacono, G.T. McCandless, D.W. Smith, B.M. Novak, Chem. Commun. 51, 9220 (2015)
G.J. Ehlert, Y. Lin, H.A. Sodano, Carbon 49, 4246 (2011)
F. Cuiqin, W. Jinxian, W. Julin, Z. Tao, Appl. Surf. Sci. 356, 9 (2015)
S.A. Mirji, S.B. Halligudi, D.P. Sawant, K.R. Patil, A.B. Gaikwad, S.D. Pradhan, Colloids Surf. A 272, 220 (2006)
K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquerol, T. Siemieniewska, K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquerol, T. Siemieniewska, Handbook of Heterogeneous Catalysis (Wiley, Weinheim, Germany, 2008)
F. Meder, T. Daberkow, L. Treccani, M. Wilhelm, M. Schowalter, A. Rosenauer, L. Mädler, K. Rezwan, Acta Biomater. 8, 1221 (2012)
A. Katiyar, L. Ji, P. Smirniotis, N.G. Pinto, J. Chromatogr. A 1069, 119 (2005)
J. He, Z. Liu, C. Hai, AIChE J. 54, 2495 (2008)
A.L.-T. Pham, D.L. Sedlak, F.M. Doyle, Appl. Catal. B 126, 258 (2012)
S.M.L. dos Santos, K.A.B. Nogueira, M. de Souza Gama, J.D.F. Lima, I.J. da Silva Jr., D.C.S. de Azevedo, Microporous Mesoporous Mater. 180, 284 (2013)
G. McKay, J. Chem. Technol. Biotechnol. 32, 759 (2007)
Y. Al-Degs, M.A.M. Khraisheh, S.J. Allen, M.N. Ahmad, Water Res. 34, 927 (2000)
F. Felsovalyi, P. Mangiagalli, C. Bureau, S.K. Kumar, S. Banta, Langmuir 27, 11873 (2011)
L. Ye, C. Liang, J. He, J. Ou, Q. Wu, J. Inorg. Organomet. Polym. Mater. 28, 1011 (2018)
M. Ferenc, N. Katir, K. Milowska, M. Bousmina, Y. Brahmi, A. Felczak, K. Lisowska, M. Bryszewska, A. El Kadib, Microporous Mesoporous Mater. 231, 47 (2016)
B. Saha, J. Saikia, G. Das, RSC Adv. 3, 7867 (2013)
K. Kubiak-Ossowska, K. Tokarczyk, B. Jachimska, P.A. Mulheran, J. Phys. Chem. B 121, 3975 (2017)
L. Ji, A. Katiyar, N.G. Pinto, M. Jaroniec, P.G. Smirniotis, Microporous Mesoporous Mater. 75, 221 (2004)
S.R. Durell, B.R. Brooks, A. Ben-Naim, J. Phys. Chem. 98, 2198 (1994)
S.T. Moerz, P. Huber, J. Phys. Chem. C 119, 27072 (2015)
Acknowledgements
This research was supported by the Polish National Science Centre under Grant No. DEC-2012/05/D/ST5/03488 titled Synthesis and characterization of modified organosilica sorbents designed for the sorption of biomolecules. The XRD characterization was carried out with the equipment purchased owing to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-06-024/09 Center of Functional Nanomaterials). M.Sc. Emil Zięba from John Paul II Catholic University of Lublin is acknowledged for SEM and TEM characterization.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Barczak, M. Functionalization of mesoporous silica surface with carboxylic groups by Meldrum’s acid and its application for sorption of proteins. J Porous Mater 26, 291–300 (2019). https://doi.org/10.1007/s10934-018-0655-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-018-0655-7