
Abstract
The extensional viscosity is an important rheological characteristic of polymer melts. It is however not as frequently reported on as the shear viscosity. The extensional viscosity is of special interest when considering polymeric materials for foaming and film blowing processes. Here, the extensional (and shear) viscosity along with the melt strength and the tensile properties of the corresponding solid film of a β-glucan concentrate are reported on. A capillary viscometer equipped with a hyperbolic die, yielding a contraction flow, was used to assess the extensional viscosity of the aqueous β-glucan compound at room temperature and at elevated temperatures (110 and 130 °C). In general, the extensional viscosity as well as the shear viscosity decreased with increasing deformation rate. The influence of two different amounts of added water (40 and 50%) was also examined. As expected, both types of viscosities decreased with increasing temperature. It is suggested that gelatinization of the starch fraction in the concentrate at 110 and 130 °C contributes to temperature dependence of the viscosity. To some extent, this is supported by light microscopy and confocal scanning laser microscopy studies of the microstructure of the materials. The results reported here indicate that the β-glucan concentrate might, after some modifications, be used as a complement to fossil-based polymers and processed by conventional manufacturing techniques.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extrusion, or hot-melt extrusion, is one of the most important methods within plastics processing. It is a versatile, continuous process, and several items such as cables, tubes and coatings as well as foamed articles are often produced by this technique. In order to control the properties of the produced items, it is important to define the specific processing parameters, but also to have an understanding of the rheological behaviour of the polymer melt during the process. For foamed articles, the extensional or elongational viscosity, together with the melt strength, becomes of special interest since the ability of the polymeric material to sustain a biaxial and extensional flow around a growing gas-bubble is a crucial property for foaming. The importance of extensional rheology for foaming has been highlighted e.g. in the work of Xu et al. and Stange and Münstedt for polypropylene [1, 2]. Furthermore, Zoller and Marcilla studied the foaming behaviour of plasticized poly(vinyl chloride-co-vinyl acetate) and found the extensional viscosity of the melt to be an important property with regard to the foaming ability [3]. Foams made of low density polyethylene (LDPE) using two different blowing agents were studied by Ramesh et al. They concluded that a higher extensional viscosity gave a higher melt strength, which was considered as favourable for foaming. It was also noted that addition of blowing agents to the LDPE reduced the extensional viscosity at a given strain rate [4].
Even though the extensional viscosity is believed to be one of the more critical rheological properties, together with an associated strain hardening, during extrusion foaming, there is still a need for more detailed studies of its importance in material processing and its relation to material characteristics. A difficulty in this context is a limitation in the number of available, reliable, measuring methods and the experimental difficulties associated with these techniques [5]. Much of the early experimental work on extensional viscosity was performed by Meissner [6] and Münstedt [7, 8] and techniques based on opposing jets, filament stretching, squeeze flow, melt extension, four-roll milling etc. have been suggested and analysed, cf also [5, 9]. Each measuring system has its advantages and limitations. For mobile liquids, like polymer melts and polymer solutions, the extensional viscosity can be assessed by subjecting the fluid to a contraction flow in a hyperbolic nozzle or die. The nozzle is then designed to give a constant extension rate and by measuring the required pressure drop, the extensional viscosity can be estimated [10, 11]. Contraction flow was used in this work and a capillary viscometer was used for both the shear and the extensional measurements.
A twin-bore capillary viscometer, equipped with a constant diameter capillary and an orifice die (Cogswell’s method), has previously been used to obtain the shear and extensional viscosity of hydroxypropyl cellulose (HPC) [12]. The HPC was found to be strain-thinning and exhibited extensional viscosities in the range of about 1000–100 kPa s at extension rates from about 2–200 s−1. A similar procedure using the Cogswell method was used by Pitts et al. to study rheological properties, mainly in shear, of wheat and corn blends [13]. Padmanabhan and Macosko investigated the extensional viscosity of LDPE by using a capillary viscometer equipped with an orifice and a regular die and comparing the result of using the entrance pressure drop method with a fibre wind-up method [14]. It was concluded that the results from the Cogswell method were in good agreement with those from the fibre wind-up technique. Furthermore, for biopolymers, the contraction flow technique has been used to demonstrate the importance of extensional flow properties of gluten and other cereal prolamins and dispersions of nanocellulose by e.g. [15,16,17].
It is obvious that knowledge of not only the shear, but also of the extensional rheological properties is of great importance for processing of materials. It further becomes of special interest when dealing with “new materials”, such as bioplastics or biopolymers, for which it is important to assess to which extent the differences in properties compared with conventional plastics needs to be considered during processing. One such polymeric material, currently being explored for its potential as a substitute for some fossil-based polymers, can be found within the agricultural sector. β-Glucan is a hemicellulose, and the β-glucan concentrate used here is often referred to as a dietary fibre. This hemicellulose is the most common non-starch component in the cell wall of some cereal grains [18]. It is particularly abundant in oat and barley but can also be produced by yeast and mushrooms. β-Glucan consists of glucose chains, connected by β-(1→3) and β-(1→4)-linkages. The introduction of the β-(1→3)-linkages in the polymeric β-(1→4)-chain makes the polymer irregular, hindering uniform packing of the polymers and thus makes the material water soluble. Despite that several studies have shown that β-glucan may play an important role in lowering of the low-density lipoprotein (LDL) cholesterol [19,20,21], most of the barley β-glucan still goes into animal feed whereas the oat β-glucan is more common as additive in human food products [18]. The β-glucan concentrate used here originated from barley.
Experimental
Material
The β-glucan concentrate was kindly supplied by Lyckeby Starch AB (Sweden). The product originated from barley and the composition, as given by the supplier, is given in Table 1. Commercially, the product goes by the trade name: Lyckeby beta-glucan fiber DF 25.
The dietary fibre fraction consists of β-glucan, arabinoxylan, cellulose and some proteins.
Methods
Preparation of Material
The β-glucan concentrate in powder form was mixed with water for 5 min using a temperature-controlled mixing chamber (Brabender, Germany) with a volume of 50 cm3. The added amount of water was 40 or 50% by weight and the actual water content was determined gravimetrically after the mixing.
Evaluating the Shear and the Extensional Viscosities
A capillary viscometer (Göttfert Rheograph 2002, Germany) was used to assess the rheological properties of the material. The shear viscosity was determined as a function of the shear rate in the conventional manner by measuring the pressure required to force the melt through a capillary at a certain piston speed. The piston speed was varied in order to obtain a series of shear rates. The cylindrical capillary used had a length of 10 mm and a diameter of 1 mm.
Here, the extensional viscosity was obtained by replacing the regular die, being a cylindrical capillary, with an axisymmetric hyperbolically shaped die during the measurements. This hyperbolic die had a length of 20 mm, an inlet diameter of 3 mm and an outlet diameter of 1 mm. Subsequent calculations based on the hyperbolic contraction flow theory gave the extensional viscosity (as described later).
The regular cylindrical capillary was used to measure the shear viscosity of the material and the hyperbolic die to evaluate the extensional viscosity. The use of contraction flow for assessing the extensional viscosity is based on a model developed by Binding [10] and later applied by e.g. [15, 22, 23]. The rheological properties were determined at three different temperatures; 30, 110 and 130 °C. A 50 or a 500 bar pressure transducer (Dynisco, USA) was used to record the pressure during the measurements. The β-glucan concentrate, mixed with water (40 or 50%) was transferred into the barrel (diameter 12 mm) above the capillary at room temperature. The material was heated in the barrel to the set temperature (30, 110 or 130 °C) and then forced through the die at different piston speeds, corresponding to different shear rates. Piston speeds of 0.001–2 mm s−1 were used, giving shear rates in the region of 1.8–3600 s−1 (in case of the cylindrical capillary). A summary of the experimental conditions used is shown in Tables 2 and 3. In the present study, the measured values were not subjected to the Rabinowitsch- or the Bagley-corrections.
The experimental values obtained with the regular die (cylindrical capillary) were fitted to a power-law relation, i.e.,
where ɳs is the shear viscosity, ɣ̇ is the shear rate, K is the consistency index and n is the power-law index. At least three measurements were carried out for each sample and an average of the three measurements were used for the extraction of K and n.
The measured pressure drop over the hyperbolic nozzle will be partly affected by shear flow from wall interactions in addition to the pressure associated with the extensional deformation. For this reason, a correction must be made for the shear flow contribution. For a power law fluid, this contribution can be calculated using [15]:
where H is the length of the die, Q is the volume flow rate and r0 and r1 are the entrance and exit radii of the die, respectively.
The extensional strain rate ε̇ was calculated from the results obtained with the hyperbolic die using
The extensional viscosity is then obtained as
where pm is the measured pressure drop over the hyperbolic die and ps is the calculated shear contribution from Eq. (2).
Melt Strength
The melt strength and the corresponding elongation at rupture was measured with a Rheoscope 1000 CEAST 6742/000 (Ceast SpA, Italy) capillary viscometer. This viscometer is equipped with a rotating disc unit (positioned below the exit of the capillary) which picks up the extruded strand from the capillary, stretches it, giving the stress–strain response of the compound. The strain in the strand was evaluated from the difference between the tangential velocity of the rotating disc and the velocity of the extruded strand at the exit of capillary, the latter being kept at 15 mm s−1. During the experiments, the velocity of the rotating disc was linearly increased at a rate of 0.63 mm s−2, thus stretching the extruded strand. The force required for extension of the melt was measured with a transducer. The initial temperature of the compounds was kept at about 20 °C. The measurements are carried out in a non-isothermal state, but this is not expected to affect the conclusions to any larger extent. The β-glucan concentrate was compounded with 40% water at 90 °C in a temperature-controlled mixing chamber (Brabender, Germany) at 30 rpm for 5 min and was let to cool in a plastic zip-lock bag before the material was transferred to the capillary viscometer. The average values of the melt strength and elongation at rupture from three to five separate measurements are reported.
Microscopy
For examination of the microstructure of the β-glucan concentrate mixed with water (50% added water) with light microscopy (LM) and confocal laser scanning microscopy (CLSM), the samples were rapidly frozen in liquid nitrogen and transferred to a cryostat (Leica CM 1900, Wetzlar, Germany) for sectioning (10 µm) at − 15 °C. For the optical microscopy, the sections were stained with Light Green (0.1% in acetic acid) and Lugols solution and examined under a light microscope, (Nikon Microphot-FXA, Tokyo, Japan). Images were taken with a NikonFX-35DX camera and processed using CellSens Dimention (Olympus, Japan).
In order to visualize the β-glucan, the sections were stained with Calcofluor and Texas Red® (protein), and analysed in a Leica TCS SP2 (Leica Ltd, Heidelberg, Germany) confocal laser scanning microscope.
Preparation of Free-Standing Films
Films for the tensile testing were prepared by mixing 3% by weight of the β-glucan concentrate with deionised water at 95 °C, using a magnetic stirrer, for about 5 min. The temperature of 95 °C was chosen in order to ensure that the starch fraction in the concentrate was properly gelatinized. Even though barley starch has a peak gelatinization temperature around 55–65 °C [24,25,26], the higher temperature of 95 °C, which is common when gelatinising potato starch [27], was used in order to ascertain that the crystalline starch structure was disintegrated and the material transformed to a molecular solution. The films were casted in 85 mm diameter Petri dishes and were dried at room temperature for about 5 days.
Tensile Testing
Tensile tests were carried out using an Instron 5542 Universal Testing Instrument (Instron Corporation, Canton, USA). Free-standing films, prepared as described above, were conditioned for a minimum of 2 days at 20 °C and 55% relative humidity prior to testing. Rectangular tensile specimens, with dimensions of 20 × 6 × 0.5 mm3 were cut out from these films, clamped and drawn at room temperature at a constant rate of 0.05 mm s−1. It is assumed that no significant drying occurred during the short test time. For each sample, three separate measurements were performed and average values of the tensile modulus, tensile strength and elongation at rupture were calculated. Three separate measurements on this compound were performed and the tensile strength and elongation at rupture were evaluated.
Results
Moisture Content
Without any added water, the β-glucan concentrate had an initial water content of about 7% (at room temperature in equilibrium). The measured water content after mixing was then often higher than what corresponded to the amount of added water. For example, when 50% water was added and the material was mixed at 90 °C, the measured water content afterwards was about 52%. There was a slight difference between mixing at 90 and 30 °C, but not large enough to be considered as important; i.e-the water contents differed by less than 1%.
Shear Viscosity of the β-Glucan Concentrates
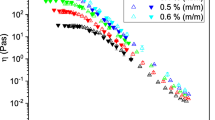
The shear viscosity was measured as a function of the shear rate with the capillary viscometer using a regular 180° circular die with a diameter of 1 mm and a length of 10 mm. The results are shown in Fig. 1 and the values shown are averages from at least three measurements.

The shear viscosity as a function of the shear rate for the nine different compounds, corresponding to experiments R1–R9
All the nine different β-glucan concentrates exhibited a shear-thinning behaviour, i.e. decreasing viscosity with increasing shear rate, as is common for polymeric melts. The highest viscosity was noted for compound R7, which contained the lower amount of water (40%), was mixed at 90 °C and measured at 30 °C. This is not surprising since lower water or plasticiser content should give a higher viscosity (as well as a lower temperature of the material). The lowest viscosity was obtained with compound R2 (although R4 and R9 exhibited viscosities in the same range). These compounds were similar in that they all contained more water (50% added) and the measuring temperature was above the gelation temperature of the starch material (110 and 130 °C). A distinct division of two groups of compounds with more similar viscosities can be seen in Fig. 1. What distinguishes the two groups is the temperature at which the viscosity was determined, which is not very surprising. If the temperature of the suspension was 30 °C (below the gelatinization temperature of the starch component), this was reflected in a higher viscosity, whereas a temperature above the gelatinization temperature, i.e. 110 or 130 °C, lower viscosities were measured. For the compound with the highest viscosity (R7), viscosities in the range of 3 × 104–340 Pa s were obtained at shear rates ranging from 1.8 to 1440 s−1. In the case of the compound with the lowest viscosity (R2), viscosities in the range of 99–8 Pa s were noted at shear rates ranging from 90 to 5400 s−1.
Within each of the two groups, there also appear to be a certain pattern. A higher water content led to a lower viscosity (which is to be expected), whereas a higher mixing temperature resulted in a somewhat higher viscosity level. However, regarding the shear viscosity, the temperature of the suspension seems to be more important than these two other conditions (at least within the ranges used here).
The material parameters, the consistency K and the flow index n, were obtained by fitting the shear data to the power law, Eq. (1), although it is obvious that the power law does not provide a perfect description of the shear flow. The parameters for the different compounds are given in Table 4.
In general, the flow index n attained values around 0.40 (with exception of compound R1), which is not very different from that of synthetic thermoplastics at relevant melt temperatures, cf e.g. [28].
Extensional Viscosity
The extensional viscosity was evaluated from Eqs. (3) and (4) by measuring the pressure drop over the hyperbolic die in the capillary viscometer and then subtracting the contribution from the shear flow. Figure 2 shows the calculated extensional viscosity as a function of the extension rate for the other compounds. The result for compound H8 was excluded due to difficulties in obtaining a stable pressure reading and thus the measurement was not considered to be reliable.

The extensional viscosity as a function of the extension rate for the compounds H1–H9 (excluding H8)
The highest extensional viscosity was obtained for compounds H5 and H7. Both compounds had the lower water content of 40% and the measurements were performed at 30 °C. A similar trend as for the shear viscosity can thus be observed. The lowest extensional viscosities were noted with compounds H2 and H9. These compounds were similar in that they contained higher amounts of water (50% added) and the measurement temperatures exceeded the gel point of starch (110 and 130 °C). Here, a similar division into two groups of compounds with more similar viscosities can be seen also for the extensional viscosity and the reason is believed to be the same as in the case of the shear viscosity. For the compounds with the highest viscosities (H5 and H7), extensional viscosities in the range of 1.6 × 108–3.1 × 107 Pa s were obtained at extension rates ranging from 1.18 × 10−2 to 1.18 × 10−1 s−1 and 4.9 × 107 to 2.5 × 106 Pa s at extension rates of 1.1 × 10−1–8.8 s−1, respectively. For the compounds with the lowest viscosities (H2 and H9), extensional viscosities in the range of 2.6 × 105–3.7 × 104 Pa s were obtained at extension rates ranging from 1.1 to 22.6 s−1 and 2.6 × 105 to 4.8 × 104 Pa s at extension rates of 1.16–23.2 s−1, respectively.
Melt Strength of β-Glucan Concentrate Filaments
Typically, about ten measurements were carried out for the material compound on different occasions and from those about 3–5 rheological measurements were considered reliable. The average melt strength of the considered measurements was about 55 mN and the experimental scatter in the melt strength results was in most cases around 10% or lower. The measured values of the strain at failure exhibited an appreciable scatter, which perhaps not is unexpected since the result is sensitive towards local disturbances. In the present case, drying of the material surface or regions with unevenly mixed material are examples of such disturbances. The average strain was about 65%. However, the relative experimental scatter was of the order of 25% (expressed in relative terms). It may be noted that attempts to measure the stress–strain behaviour of the compounds at higher temperatures were unsuccessful, since the compounds simply became too weak to yield any significant force reading, i.e. the melt strength became very low.
As a comparison, Thunwall et al. measured the stress–strain properties of plasticized starch melts at elevated temperatures (120–140 °C) [29]. For these starch-based materials, the melt strength was in many cases of the same order as for the β-glucan concentrates used here, but the elongation at failure was at least twice as high (or significantly higher). The difference in processing temperature and plasticizer content should however be noted.
Microscopy
Four different samples, at different processing stages, of the mixed β-glucan concentrate, 50% added water, were taken for microstructural analysis using LM. In Fig. 3a and c the concentrate was mixed with the added water at 90 °C and 30 °C, respectively, for 5 min in the mixing chamber. In Fig. 3b and d the samples had been further processed in the capillary viscometer at 130 °C. The purple–brown colour represents starch, the green colour represents protein and the white parts are fibres (containing β-glucan). In Fig. 3c, the intact starch granules (brown–violet particles) are clearly visible since the temperature did not exceed 30 °C. However, already after mixing at a temperature of 90 °C, the granules were to some extent disrupted/melted and transformed into a continuous phase. As can be seen in Fig. 3b and d, the starch is evenly distributed in the structure and forms in a sense a continuous phase when the material has been processed in the capillary viscometer at 130 °C, regardless of any initial difference after the mixing step at different temperatures. It therefore seems unnecessary to compound the material with added water at the higher temperature, i.e. a lower temperature will lead to energy savings and provide an easier preparation of the compound.

Optical micrographs of (a) β-glucan concentrate mixed with 50% water at 90 °C. b β-Glucan concentrate mixed with 50% water at 90 °C and processed with the capillary viscometer at 130°. c β-Glucan concentrate mixed with 50% water at 30 °C. d β-Glucan concentrate mixed with 50% water at 30 °C and processed with the capillary viscometer at 130 °C. Purple–brown colour denotes starch, green colour denotes protein and white colour the fibres. (Color figure online)
The β-glucan is located within the fibres, i.e. in the unstained white parts in the structures shown in the LM-images in Fig. 3. (Some plant cell wall fractures are also seen as weakly green since the cell walls also contain protein.) β-Glucan can be fluorescently stained and shown specifically using the CLSM. Figure 4 provides an example in case of a compound that had been mixed with water at 90 °C. The β-glucan, which is shown in blue, is in the fibrous parts of the structure. Other parts of the cell wall, fibres and proteins are coloured in red.

CLSM images of a β-glucan–water compound produced by mixing at 90 °C. The β-glucan is shown in blue, and other fibre parts and protein appear in red. (Color figure online)
Tensile Properties of Films of the β-Glucan Concentrate
The tensile testing of the solid β-glucan concentrate was performed in triplicate in order to obtain an estimate of the experimental scatter. The test bars were strips cut out from the casted films, which were produced as described in the “Experimental” section. It is here assumed that the moisture content of the films was about 7%. Figure 5 shows the corresponding stress–strain curves. As can be seen, the three measurements were fairly consistent. A tensile stress of about 25–30 MPa was reached before failure and the corresponding elongation at rupture was about 1%. The films were apparently quite brittle in the solid state. Zárate-Ramírez et al. investigated the mechanical properties of a gluten-based biopolymer and evaluated the effect of adding plasticiser and three different kinds of polysaccharides to the compound. With these materials, they found a maximum tensile strength of about 2.7 MPa and an elongation at break of about 180% [30]. This would indicate that the β-glucan films are relatively strong and brittle in comparison to the gluten-based systems. Thunwall et al. investigated the mechanical properties of film-blown thermoplastic starch (containing glycerol as plasticizer). The tensile properties of such starch films will depend strongly on the plasticizer content, but it may be noted that in that study the reported tensile strength was lower than that of the β-glucan films used here, but the strain at failure was appreciably higher; of the order of 10% [31].

Stress–strain curves (measured in triplicate) for solid films of the β-glucan concentrates
Discussion
At present, there is a general strive for replacing fossil-based polymers with suitable biopolymers or other types of biomaterials, for example cellulose- or starch-containing compounds, in different applications. Such applications may include e.g. packaging materials, structural elements in furniture and interior components in automotive industry. A prerequisite for a successful replacement is that the intended bio-based material can be processed into products in an effective manner using currently used processing techniques such as extrusion and injection moulding. This in turn calls for an understanding and characterization of the flow (rheological) properties of the bio-based material and their impact on the intended manufacturing technique.
The aqueous suspensions of the β-glucan concentrate appear to be an interesting system in this context. The flow properties of the aqueous compound resemble in a sense those of fibre-filled melts. The shear rate dependence of the shear viscosity is of the same type as for e.g. cellulose-containing polymer melts and thermoplastic starch, cf e.g. [28, 31].
The extensional properties of the β-glucan-containing system may be of special concern since these are of importance in relation to manufacturing techniques like extrusion foaming and film blowing. In a subsequent paper from our laboratories the foaming ability of aqueous β-glucan containing compounds will be described in relation to processing conditions, water content and the shear and extensional properties of the system. From the results shown here it is obvious that the concentrate is both shear and extension thinning (Figs. 1, 2) and that the Trouton ratio clearly exceeds 3, i.e. the compounds exhibit a viscoelastic character. Regarding the magnitude of the extensional viscosity of the β-glucan concentrates, it covers quite a broad range depending on the water content and treatment. It is however interesting to note that this range is quite similar to what has been reported for LDPE [32], HPC [12], prolamins [16], zein-starch compounds [33] and polyethylene and polystyrene [34], but higher than that noted for suspensions of cellulose nanofibrils [17].
As already noted, the viscosity curves in Figs. 1 and 2 fall into two groups, one for which the measuring temperature was kept at 30 °C and the other corresponded to measurements performed at higher temperatures (110 or 130 °C). The compounds in the first group exhibited the highest viscosities, which is not unexpected considering the lower measuring temperature. It is however suggested that also the change in microstructure as shown in Fig. 3, i.e. the gelatinization and homogeneous distribution of the starch component plays a role with regard to the viscosity level. The gelatinization of the starch might have positive effects on the processability of the β-glucan compound as well as on the foaming ability during extrusion. It is rather well known that starch-containing compounds can be foamed in an extrusion process [35, 36]. Extrusion foaming of the β-glucan concentrate will, as mentioned, be described in separate work.
From the LM images it is clear that the β-glucan concentrate has two structurally different phases. Depending on temperature, the concentrate is either protein- or starch continuous, Fig. 3a and c. It is interesting that the thermal history of the concentrate seems to be insignificant, when the experimental temperature exceeds the previous treatment temperature, Fig. 3b and d. This material property could be useful in order to minimize the pre-treatment steps at elevated temperatures needed.
The tensile testing of the β-glucan films suggested that the material is quite hard and brittle at room temperature and humidity. In certain applications these properties will be desired, however, to produce a material with absorbing or cushioning properties, a different plasticizer which cannot evaporate from the material might be of interest.
Conclusions
Aqueous suspensions of the β-glucan concentrate appear to possess rheological properties which can open up possibilities to process such compounds into useful products using conventional plastic manufacturing methods. The shear viscosity as well as the extensional viscosity seem to be in the same range as reported for e.g. fossil-based, synthetic polymers. The extensional viscosity was here assessed using the contraction flow technique which appears to be adequate for compounds of this kind. Both types of viscosities depended, as expected, on the solids content of the suspension, the processing temperature and the preparation of the suspensions. Processing at elevated temperatures also induced a gelatinization of the starch component, i.e. a change in the microstructure, of the concentrate which believed to affect the properties and the flow behaviour of the material. Tensile tests revealed however that the concentrate, when in the solid state, was quite brittle and in this respect improvements might be called for.
References
Xu Z, Zhang Z, Guan Y, Wei D, Zheng A (2013) Investigation of extensional rheological behaviors of polypropylene for foaming. J Cell Plast 49(4):317–334
Stange J, Münstedt H (2006) Rheological properties and foaming behavior of polypropylenes with different molecular structures. J Rheol 50(6):907–923
Zoller A, Marcilla A (2013) Extensional viscosity measurements and characterization of poly(vinyl chloride-co-vinyl acetate) plastisols and foams. J Appl Polym Sci 128(1):354–362
Ramesh NS, Lee ST, Lee K (2003) Novel method for measuring the extensional viscosity of PE with blowing agent and its impact on foams. J Cell Plast 39(4):281–289
Gendron R, Daigneault LE (2014) 3. Rheology of thermoplastic foam extrusion process. In: Lee S, Park CB (eds) Polymeric foams: foam extrusion: principles and practice. CRC Press, Baca Raton, pp 39–81
Meissner J (1972) Development of a universal extensional rheometer for the uniaxial extension of polymer melts. Trans Soc Rheol 16:405–420
Münstedt H (1975) Viscoelasticity of polystyrene melts in tensile creep experiments. Rheol Acta 14:1077–1088
Münstedt H (1979) New universal extensional rheometer for polymer melts. Measurement on a PS sample. J Rheol 23:421–436
Münstedt H, Schwarzl F (2014) Extensional rheology. In: Deformation and flow of polymeric materials. Springer, Berlin, pp 387–418
Binding DM (1988) An approximate analysis for contraction and converging flows. J Nonnewton Fluid Mech 27(2):173–189
James DF, Chandler GM, Armour SJ (1990) A converging channel rheometer for the measurement of extensional viscosity. J Nonnewton Fluid Mech 35(2):421–443
Paradkar A, Kelly A, Coates P, York P (2009) Shear and extensional rheology of hydroxypropyl cellulose melt using capillary rheometry. J Pharm Biomed Anal 49(2):304–310
Pitts K, Favaro J, Austin P, Day L (2014) Co-effect of salt and sugar on extrusion processing, rheology, structure and fracture mechanical properties of wheat–corn blend. J Food Eng 127:58–66
Padmanabhan M, Macosko CW (1997) Extensional viscosity from entrance pressure drop measurements. Rheol Acta 36:144–151
Wikström K, Bohlin L (1999) Extensional flow studies of wheat flour dough. II. Experimental method for measurements in constant extension rate squeezing flow and application to flours varying in breadmaking performance. J Cereal Sci 29(3):227–234
Oom A, Pettersson A, Taylor JRN, Stading M (2008) Rheological properties of kafirin and zein prolamins. J Cereal Sci 47(1):109–116
Moberg T, Rigdahl M, Stading M, Levenstam Bragd E (2014) Extensional viscosity of microfibrillated cellulose suspensions. Carbohyd Polym 102(1):409–412
Ahmad A, Anjum FM, Zahoor T, Nawaz H, Dilshad SMR “(2012) Beta glucan: a valuable functional ingredient in foods. Crit Rev Food Sci Nutr 52(3):201–212
Othman RA, Moghadasian MH, Jones PJ (2011) Cholesterol-lowering effects of oat β-glucan. Nutr Rev 69(6):299–309
Wolever TMS, Gibbs AL, Brand-Miller J, Duncan AM, Hart V, Lamarche B, Tosh SM, Duss R (2011) Bioactive oat-glucan reduces LDL cholesterol in Caucasians and non-Caucasians. Nutr J 10(1):130–134
Tiwari U, Cummins E (2011) Meta-analysis of the effect of [beta]-glucan intake on blood cholesterol and glucose levels. Nutrition 27(10):1008–1016
Isaksson P, Rigdahl M, Flink P, Forsberg S (1998) Aspects on the elongational flow behaviour of coating colours. J Pulp Pap Sci 24(7):204–209
Stading M, Bohlin L (2000) Contraction flow measurements of extensional properties. Trans Nord Rheol Soc 8/9:181–185
Tester RF, Morrison WR (1990) Swelling and gelatinization of cereal starches. I. Effects of amylopectin, amylose, and lipids. Cereal Chem 67(6):551–557
Myllärinen P, Schulman AH, Salovaara H, Poutanen K (1998) The effect of growth temperature on gelatinization properties of barley starch. Acta Agric Scand Sect B 48(2):85–90
Song Y, Jane J (2000) Characterization of barley starches of waxy, normal, and high amylose varieties. Carbohyd Polym 41(4):365–377
Venkatesh A, Boldizar A (2016) Plasticizing starch by adding magnesium chloride or sodium chloride. Starch/Stärke 68:1–9
Thunwall M, Boldizar A, Rigdahl M, Banke K, Lindström T, Tufvesson H, Högman S (2008) Processing and properties of mineral-interfaced cellulose fibre composites. J Appl Polym Sci 107(2):918–929
Thunwall M, Boldizar A, Rigdahl M, Kuthanová V (2006) On the stress–strain behaviour of thermoplastic starch melts. Int J Polym Anal Charact 11(6):419–428
Zárate-Ramírez L, Romero A, Bengoechea C, Partal P, Guerrero A (2014) Thermo-mechanical and hydrophilic properties of polysaccharide/gluten-based bioplastics. Carbohyd Polym 112:24–31
Thunwall M, Kuthanová V, Boldizar A, Rigdahl M (2008) Film blowing of thermoplastic starch. Carbohyd Polym 71:583–590
Ramesh NS, Lee ST (2000) Blowing agent effect on extensional viscosity calculated from fiber spinning method for foam processing. J Cell Plast 36:374–385
Andersson H, Öhgren C, Johansson D, Kniola M, Stading M (2011) Extensional flow, viscoelasticity and baking performance of gluten-free zein-starch doughs supplemented with hydrocolloids. Food Hydrocoll 25(6):1587–1595
Laun HM, Schuch H (1989) Transient elongational viscosities and drawability of polymer melts. J Rheol 33(1):119–175
Willett JL, Shogren RL (2002) Processing and properties of extruded starch/polymer foams. Polymer 43(22):5935–5947
Sjöqvist M, Boldizar A, Rigdahl M (2010) Processing and water absorption behavior of foamed potato starch. J Cell Plast 46(6):497–517
Acknowledgements
The funding from the Swedish Research Council Formas is gratefully acknowledged. Further thanks are extended to the members of the SmartFoam project for initiating this research. Special thanks to Dr Roland Kádár, Chalmers University of Technology, for helpful discussions and assistance with the melt strength measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Karlsson, K., Berta, M., Öhgren, C. et al. Flow Behaviour and Microstructure of a β-Glucan Concentrate. J Polym Environ 26, 3352–3361 (2018). https://doi.org/10.1007/s10924-018-1207-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-018-1207-6