
Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare genetic disorder characterized by impaired immunity against intracellular pathogens, such as mycobacteria, attenuated Mycobacterium bovis-Bacillus Calmette–Guérin (BCG) vaccine strains, and environmental mycobacteria in otherwise healthy individuals. Retrospective study reviewed the clinical, immunological, and genetic characteristics of patients with MSMD in Mexico. Overall, 22 patients diagnosed with MSMD from 2006 to 2021 were enrolled: 14 males (64%) and eight females. After BCG vaccination, 12 patients (70%) developed BCG infection. Furthermore, 6 (22%) patients developed bacterial infections mainly caused by Salmonella, as what is described next in the text is fungal infections, particularly Histoplasma. Seven patients died of disseminated BCG disease. Thirteen different pathogenic variants were identified in IL12RB1 (n = 13), IFNGR1 (n = 3), and IFNGR2 (n = 1) genes. Interleukin-12Rβ1 deficiency is the leading cause of MSMD in our cohort. Morbidity and mortality were primarily due to BCG infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vaccination with attenuated Mycobacterium bovis-Bacillus Calmette–Guérin (BCG) protects infants against severe forms of tuberculosis (TB). Two different strains of the BCG vaccine have been used in Mexico: the Danish-1331 strain administered until 2005, followed by the Tokyo-172 strain in current use [1]. Adverse events following BCG immunization are among the most common manifestations of Mendelian susceptibility to mycobacterial disease (MSMD). MSMD is a rare group of inborn errors of immunity (IEI) characterized by selective susceptibility to clinical diseases caused by BCG vaccines and environmental mycobacteria in otherwise healthy patients in the absence of overt immunological abnormalities [2]. Genetic variants in 19 genes (IFNG, IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, IL12RB2, IL23R, RORC, TBX21, IRF8, SPPL2A, ISG15, USP18, TYK2, JAK1, ZNFX1, NEMO, and CYBB) lead to MSMD and define 34 disorders that reflect high levels of allelic heterogeneity [2]. The pathogenesis of MSMD depends mostly on gene variant that leads to either insufficient production or inadequate response to interferon gamma (IFN-γ), which is indispensable for an efficient immune response against mycobacterial species in humans [3]. Biallelic variants in IL12RB1 are the most frequent genetic cause, being present in approximately 60% of patients diagnosed with MSMD [4]. The severity and penetrance of MSMD are inversely correlated with residual IFN-γ activity [4]. In Mexico, only 11 cases of MSMD have been reported up to date [5,6,7,8,9,10,11,12,13,14,15,16]; the status of this IEI in different states is unknown [17]. This paper reports the genetic, immunological, and clinical features of the first cohort of MSMD patients from 10 hospitals in 4 states in Mexico.
Materials and Methods
Patients
Medical records of patients genetically confirmed as having MSMD from 2006 to 2021 in 10 hospitals in Mexico were retrospectively reviewed. This study was approved by the ethics committee of the National Institute of Pediatrics in Mexico City (Mexico). All centers were contacted via e-mail and were requested to provide patient details using a questionnaire. Demographic characteristics, family history, clinical manifestations, radiological data, treatment, and follow-up data were also obtained when available. Other information collected included routine immunological and microbiological records, laboratory functional evaluation of IFN-γ immunity, and genetic results. Clinical criteria for localized or regional (BCG-itis) and disseminated (BCG-osis) infections have been previously defined [18, 19]. Mycobacterial infection was confirmed by culture, polymerase chain reaction (PCR), histopathology, and acid-fast bacilli (AFB) findings.
Immunological Analyses
Immune evaluation of patients included phenotyping of peripheral blood lymphocyte subsets and evaluation of NADPH oxidase activity in neutrophils using dihydrorhodamine (DHR) by flow cytometry [18]. The expression of IL-12Rβ1, IFN-gR1, and IFN-gR1 was assessed by flow cytometry. The production of IFN-γ and IL-12 in whole blood after stimulation with medium alone, BCG, BCG plus IFN-γ, or BCG plus IL-12 was assessed in some patients using ELISA as previously described [7].
Genetic Analyses
Genetic diagnosis was made using Sanger sequencing (P1–P7, P12–P14, P17–22), next-generation sequencing panel (P10–P11, P16), or whole-exome sequencing (WES) (P8–P9, P15) [5,6,7,8,9,10,11,12,13,14,15]. Deleterious variants identified by WES were confirmed by Sanger sequencing. Familial segregation was performed once genomic DNA was obtained from the relatives. PolyPhen-2, which sorts intolerant from tolerant, and combined annotation-dependent depletion (CADD) scores were used to predict the pathogenic effects of the unreported variants. The Statistical Package for Social Science version 25.0 (SPSS Inc., Chicago, IL, USA) was used for data analyses. The results are presented as medians for continuous variables and as percentages for nominal variables. Survival was analyzed using the Kaplan–Meier method.
Results
Demographic Findings in 17 Kindreds of Patients with MSMD
Between 2006 and 2021, 22 patients (including 17 probands) from 17 unrelated kindreds (identified with capital letters A–P) were referred from 10 different health institutions in four states of Mexico. The country has eight geographical regions [20] and the distribution of MSMD patients according to the regions was as follows: east, n = 6; west, n = 1; north center, n = 3; south center, n = 10; and southwest, n = 2 (Table 1). Of the 22 patients, 14 (64%) were males and eight (36%) were female (Table 1). Consanguinity was positive in two kindreds. The median age at first manifestation was 6 months (range: 4 months to 22 years), and the median ages at clinical and genetic MSMD diagnosis were 4.5 years (range: 3 months to 33 years) and 8.3 years (range: 5 months to 34 years), respectively, except for two patients in whom genetic confirmation was made 4 months postmortem. The median age of the living patients at the time of the study was 14 years (range: 4–51 years).
Genetic Findings in 17 MSMD Kindreds
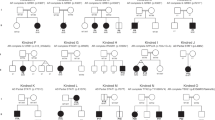
Pathogenic variants in MSMD genes were detected in all kindreds using Sanger sequencing (n = 10), copy number variants (n = 1), target gene sequencing panel (n = 3), and WES (n = 3) [5,6,7,8,9,10,11,12,13,14,15], affecting IL12RB1 in 13 kindreds (76%), IFNGR1 in 3 (18%), and IFNGR2 in one (6%) (Table 1). Biallelic variants in IL12RB1 were c.655A > T (p.S220C), c.402C > A (p.Y134*), c.517C > T (p.R173W), c.635G > A (p.R212Q), c.1456C > T (p.R486*), c.1561C > T (p.R521*), c.1750C > T (p.S584P), c.1791 + 2 T > G (p.A573Lfs*22), and deletion of exon 8 (designated △8) (Fig. 1). All variants have been previously reported [21, 22] except for c.1750C > T, a missense variant that is exceedingly rare (gnomAD exomes AF 0.000004, allele count one Latino individual) and likely pathogenic, with a CADD Phred score of 23.4 (minimum significance cutoff value for IL12RB1 at 95% CI 3.65) [23], classified as Pathogenic in ClinVar, and as VUS by ACMG criteria (http://varsome.com.) (Supplementary Figs. 1, 2, 3). The affected residue is in the Box1 motif responsible for the interaction with JAK/STAT1. Ten kindreds had homozygous pathogenic variants and three were compound heterozygous (Table 1 and Fig. 1). Three variants were identified in the IFNGR1 gene: c.201-1G > T, c.819_822del (p.N274Hfs*2), and c.805delT (p.Y269Ifs*8). The hereditary patterns are autosomal recessive (AR) in kindred N, conferring a complete deficiency. Dominant inheritance was observed in kindreds O and P. Finally, a homozygous variant, c.371C > T (p.S124F) in IFNGR2, was found in kindred Q, which was responsible for an AR-partial disease (Table 1). Familial segregation performed in biological samples available from both parents of B, C, E, G, H, J, I, L, and M kindreds confirmed their heterozygosity for all variants identified in the respective genes (IL12RB1, IFNGR1, and IFNGR2).

Family segregation of 17 kindreds with MSMD. Each kindred (K) is designated by a capital letter (A–P), and each generation is designated by a Roman numeral (I–III). The double lines connecting the parents indicate known or presumed consanguinity. An arrow indicates the probands (P); the proband number is indicated inside of the symbol. Individuals whose genetic status could not be evaluated are indicated by the symbol “?.” IL-12Rβ1 deficiency was diagnosed in kindreds A–L, AR-complete IFN-gR1 deficiency was diagnosed in kindred M, PD IFN-gR1 deficiency was diagnosed in kindreds N and O, and AR-partial IFN-gR1 was diagnosed in kindred P
Biological Findings in MSMD Patients
Only a number of IEI confer predisposition to mycobacterial infections; therefore, depending on the clinical context, a methodological assessment of available tools must rule out these [2]. The following tests performed on each of the patients were limited to their availability in the study laboratory. We assessed the samples of 14 patients using DHR or NBT assays to measure oxidative burst, which revealed normal results (Supplementary Table 3). The serum IgG, IgM, and IgA levels were measured in 11 (50%), 6 (27%), and 10 (45%) patients, respectively (Supplementary Table 3). In five patients, global lymphopenia was observed (flow cytometry detection); the other three patients had normal values (Supplementary Table 3). In 11 (50%) IL-12Rβ1 deficiency probands (P1, P2, P4, P6, P9–P14, P16), the surface expression of IL-12Rβ1 in phytohemagglutinin (PHA)-activated T cells was absent compared to healthy controls, including P10 who had a novel mutation (Supplementary Fig. 2) [5, 6, 10, 13, 15]. Additionally, 16 patients from 13 kindreds had impaired IFN-γ production due to AR IL-12Rβ1 deficiency. The AR IFN-gR1-deficient patient (P17) showed no expression of IFN-gR1 in monocytes compared to healthy controls. In autosomal dominant (AD) IFN-gR1-deficient P21, IFN-gR1 expression in monocytes was higher than that in healthy controls. In AR IFN-gR1-deficient P22, the expression of IFN-gR1 on Epstein-Barr virus-transformed B lymphoblastoid cells (EBV-B cells) was diminished compared to that in healthy controls [11].
Mycobacterial Infections in Patients with Impaired IFN-γ Production
Fourteen (87%) patients with IL-12Rβ1 deficiency were symptomatic, whereas the rest were asymptomatic (P3 and P5). All patients were vaccinated with BCG, except for P3–P5. Intradermal administration of the BCG vaccine was performed at the upper right arm. Eleven (85%) cases (P1, P2, P6–P9, P11–P13, P15, and P16) received the Tokyo-172 strain, whereas two (P10, P14) received the Danish-1331 strain. Nine vaccinated patients developed BCG disease; the median age at the first event was 6 months (range: 4–13 months). The median duration between vaccine administration and BCG infection was 5 months (range: 1–13 months). BCG infection was present in P12 (11%) as regional BCG-itis and underwent spontaneous remission. In six patients (P1, P2, P7–P9, P11), the infection evolved from BCG-itis to BCG-osis. The P6 and P10 had regional BCG-itis, which resolved in the first year of life; however, it relapsed as BCG-osis at 10 and 15 years of age, respectively (Supplementary Table 1). The first clinical manifestation was adenitis in 12 (86%) patients, its localization was axillary (n = 9, 56%), cervical (n = 2, 12%), and supraclavicular-cervical-axillary (n = 1, 6%) (Fig. 2A). BCG-osis was present in eight patients; the infected organs were the lungs (n = 6), skin and soft tissues (n = 4), liver (n = 3), spleen (n = 3), intestines (n = 1), kidneys (n = 1), brain (n = 1), and spinal cord (n = 1) (Fig. 2B). The affected lymph nodes in patients with BCG-osis were the cervical (n = 7), axillary (n = 6), mesenteric (n = 6), mediastinal (n = 4), and inguinal (n = 2) (Fig. 2C). P1 developed portal hypertension and chronic obstructive hepatopathy due to mesenteric adenomegaly compressing the portal vein (Fig. 3A). M. bovis-BCG culture was positive in all patients with BCG-osis. Five patients with BCG-osis (P1, P2, and P6–P8) were refractory to antimycobacterial treatment and died. P9 relapsed after completing the first TB drug regimen; she is currently completing the second regimen, which has already been administered for 1 year with the plan to complete 2 years. P10 received two antimycobacterial regimens, with relapse occurring after the completion of each treatment; hence, a third regimen was administered for 2 years, resulting in recovery. P11 remained hospitalized and underwent TB drug regimen (Supplementary Table 1). P8 also had M. abscessus isolated from the purulent secretion of an abscess; it was possibly secondary to primary BCG infection (Fig. 3B).

Distribution of adenitis and BCG infection in IL-12Rβ1-deficient patients. A The graphic shows the distribution of adenitis events (including these related to local BCG infection). B The graphic shows the distribution of affected organs by BCG infection. C The graphic shows the distribution of affected lymph nodes by BCG infection

Clinical and radiological findings in IL12Rβ1-deficient patients. A Multiple serosanguinous blisters in the thoracic region in P1 secondary to BCG-osis; distended abdomen due to hepatomegaly was also observed. B Purulent right axillary adenitis in an IL-12Rβ1-deficient patient (P8); M. abscess and M. bovis-BCG were isolated from purulent secretions. C A cerebral abscess on computed tomography scan. The abscess in the left frontal lobe shifts the midline in P13. Salmonella enterica group D was isolated in the purulent secretion. D Fistulized and purulent cervical adenitis developed in P14 due to Salmonella infection
BCG Infections in Patients with Impaired Response to IFN-γ
Of the 22 patients with MSMD, six had diseases with an impaired response to IFN-γ. AR-complete IFN-gR1 deficiency was identified in P17 [8], P18, and P19; AD IFN-gR1 deficiency in P20 and P21; and AR-partial IFN-gR2 deficiency in P22 [11]. P17 had two cousins (P18 and P19) with identical AR-complete IFN-gR1 deficiency. The cousins were born in the USA where BCG vaccination is not mandatory. Among the four probands (P17–P22), the first clinical manifestation was adenitis in three (75%) and multiple osteomyelitis in one (25%). All patients received intradermal BCG vaccine in the upper right arm; P17 received the Danish-1331 strain, whereas the rest received the Tokyo-172 strain. The vaccination age was before 2 months old (P17, P20, and P22) and at 2 years old (P21). P17, P21, and P22 showed adverse reactions to the BCG vaccine; P17 and P21 developed BCG-osis 1 and 3 months after vaccination, respectively, while P21 experienced two BCG-osis episodes at first year and 17 years after vaccination. Additionally, P22 had BCG-itis 2 months after vaccination. According to the method of diagnosis, M. bovis-BCG was isolated from the bronchoalveolar lavage of P17. For P21, the diagnosis of the first event was based on the clinical and radiological findings, whereas the second was based on a positive M. bovis-BCG culture. For P22, the diagnosis of BCG-itis was based on the clinical findings and disease resolution following 6 months of isoniazid (H) treatment. P17 had bone, skin, and lung as infected organs and her complete resolution was not achieved due to inadequate adherence to TB drug regimen. In P21, the first infection affected multiple lymph nodes but improved after completing a 9-month regimen of isoniazid (H), pyrazinamide (Z), and rifampicin (R); however, the second event involved the lungs, liver, spleen, lymph nodes, and bones. In P22, BCG-itis resolved after 6 months of H treatment.
Other Mycobacterial Infections in Patients with Impaired Response to IFN-γ
There is a common pathogenic mechanism to all MSMD disorders: impairment of the production of or responses to IFN-γ [4]. Patients of the first group are more severely affected [24]. P20 had two different episodes of mycobacterial infections as documented by histopathological findings. At 4 years old, the patient developed a disseminated infection affecting the liver, spleen, multiple bones, and lymph nodes that resolved after an intensive 3-year TB drug regimen. The patient’s second episode occurred at 7 years old and affected multiple bones, lungs, chest wall, and soft tissue, resulting in multi-organ dysfunction and death [14]. At 9 years old, P21 had disseminated M. tuberculosis complex infection (vertebrae, lymph nodes, and liver) detected by PCR and resolved with an antimycobacterial regime. However, at 26 years old, he developed disseminated M. colombiense infectious disease (lung, spleen, multiple lymph nodes, and vertebrae) that responded poorly to a TB drug regimen. At 28 years old, he developed disseminated M. avium infection in the intestines, spleen, lungs, vertebrae, and lymph nodes and is currently being treated with clarithromycin, R, and ethambutol (E). At 2 years old, P22 had clinical, histological, and radiological lymphatic and pulmonary TB that improved after treatment with H, R, and Z for 18 months. At 9 years old, he developed peritoneal and cutaneous mycobacterial infections (erythema nodosum), and a skin biopsy revealed AFB. The infection was ameliorated by a 9-month treatment with H, R, and Z. One year later, the patient developed cerebral TB caused by M. tuberculosis complex and confirmed by PCR; treatment involved H, R, Z, and E. At 18 years old, the patient developed disseminated mycobacterial infection in the lungs and skin, and the culture was positive for M. avium. Treatment consisted of a 1-year regimen of H, R, E, and clarithromycin; however, a relapse at 23 years old may have been due to poor adherence to treatment (Supplementary Table 1).
Salmonella and Other Bacterial Infections in Patients with MSMD
Approximately half of the patients with MSMD, particularly those with IL-12Rβ1 or IL-12p40 deficiency, are also susceptible to non-typhoidal Salmonella infections, ranging from gastroenteritis to sepsis [4, 7, 25]. In this cohort, six patients (22%) had Salmonella infection, including five with AR-complete IL-12Rβ1 [26] deficiency (P7, P9, P12, P13, and P14) and one with AR-complete IFN-gR1 deficiency (P17) (Supplementary Tables 1 and 2). P7 had one isolate of non-typhoidal Salmonella in the bone marrow culture. At 3 years old, P9 was diagnosed with Henoch-Schönlein purpura with arthralgias and reddish-purple spots in the abdomen and lower extremities. Microorganism isolation was negative; however, the patient responded well to antibiotic treatment. P12 had 30 episodes of septicemia caused by Salmonella group B, S. enterica serotype Typhi, S. choleraesius, and S. enteritidis, including recurrent Henoch–Schönlein purpura and arthritis, with renal biopsy showing IgA nephritis [12]. Considering a possible Salmonella carrier state [27], a cholecystectomy was performed; however, this did not improve the patient’s condition. Monthly administration of immunomodulatory dose of subcutaneous immunoglobulin [28] improved the patient’s condition. P13 had five Salmonella enterica group D infections from 18 months to 5 years old that were diagnosed using blood, stool, and brain abscess secretion cultures (Fig. 3C). She required drainage of the abscess due to intracranial hypertension. At 2 years old, the patient developed Henoch–Schönlein purpura. At 1 year old, P14 had a localized infection that featured giant, fistulized, and purulent cervical adenitis (Fig. 3D). The Salmonella grew in lymph node pus secretion and recurrence occurred despite antibiotic treatment. The patient was ultimately treated with a specific dialyzable leukocyte extract with no recurrence of the infection. P17 experienced septic shock due to Salmonella spp. and required intensive therapy. Meanwhile, P7, P11, and P16 were the first to contract mycobacterial infections. Secondary bacterial infections developed such as sepsis by K. pneumoniae for P7, Acinetobacter ursingii and Stenotrophomonas maltophilia for P11, and Pseudomonas aeruginosa for P16. The P7 deceased and the P11 and P16 ameliorated with intravenous broad-spectrum antibiotics with no complications.
Fungal Infectious Diseases in Patients with MSMD
Patients with MSMD are also susceptible to fungal infections, such as Candida, Paracoccidioidomyces, and Histoplasma (http://varsome.com) [7, 24,25,26]. Overall, 11 (50%) MSMD patients had at least one fungal infection; ten patients had IL-12Rβ1 deficiency; and one had IFN-gR1 deficiency (Supplementary Tables 1 and 2). Candida infections developed in eight patients (31%; P1, P2, P4 [16], P6, P7, P9, P10, and P11); each patient had at least one episode. According to the site of infection, mucocutaneous candidiasis was present in six (75%) patients, urinary tract in three (38%), gastric juice in three (38%), and central nervous system in one (13%). Furthermore, three (14%) patients with AR-complete IL-12Rβ1 deficiency (P12, P15, and P16) from regions with a humid subtropical climate developed disseminated infections caused by Histoplasma spp., particularly affecting the lymph nodes. P12 had two histoplasmosis events; the first was identified in a lymph node biopsy at 3 years old, while the second was found in the bone marrow at 4 years old. Remission was achieved in both episodes with antifungal treatment. At 20 years old, P15 contracted disseminated histoplasmosis affecting the lungs, liver, spleen, and multiple lymph nodes. Remission was achieved with amphotericin 5 mg/kg/d for 30 days, and the patient is currently maintained on itraconazole 200 mg twice a day until date. At 6 years old, P16 contracted a disseminated histoplasmosis infection that affected the lymph nodes, liver, and spleen and was treated with amphotericin B 5 mg/kg/d for 32 days. Four years later, disseminated histoplasmosis recurred and affected the meninges, lymph nodes, liver, spleen, and bone marrow. Splenectomy was necessary because of the presence of multiple fungal abscesses, and the infection was again treated with amphotericin B 5 mg/kg/d for 42 days. The patient subsequently developed hemophagocytic syndrome and demyelinating axonal neuropathy of the left limb and is currently maintained on itraconazole 200 mg daily until date.
Viral Infectious Diseases in MSMD Patients
Patients with MSMD do not have a selective predisposition to severe viral infections [24, 26, 29, 30]. In this cohort, five (23%) patients had a viral infection (P9, P11, P20, P21, and P22) (Supplementary Tables 1 and 2). Interestingly, P9 had non-complicated varicella and three viral pneumonia events. P9 was first hospitalized at 3 years old and required oxygen for respiratory syncytial virus pneumonia, which improved without complications. At 5 years old, she developed influenza A (H1N1) pdm09 pneumonia, which required mechanical ventilation. Months later, she was readmitted because of severe acute respiratory syndrome–related coronavirus-2 (SARS-CoV-2) pneumonia and was treated with supplemental oxygen with satisfactory resolution. P11 had SARS-CoV-2 pneumonia at 4 years old that required supplemental oxygen. P20 developed a systemic EBV infection associated with hemophagocytic syndrome, which improved with the HLH-04 chemotherapy protocol [31] that included etoposide, dexamethasone, and cyclosporine A. At 15 years old, P21 had a herpes zoster virus infection in the right upper limb without relapse. Finally, P22 was infected with SARS-CoV-2, which resolved with ambulatory treatment. Although viral infections occurred in some of the patients, all resolved successfully. The data from this cohort confirmed previous viral features in MSMD.
Mortality and Survival in MSMD Patients
The mortality of patients with MSMD varies depending on the genetic defects and clinical outcomes, infectious agents, and therapeutic approaches [7, 24]. Among the 22 patients, 20 were followed up at the hospital or death. After confirmation of MSMD, the median follow-up time was 4 years (range: 6 months–17 years); 7 of the 22 patients died (P1, P2, P6, P7, P8, P17, and P20). The global survival rates of patients with MSMD are 92% and 83% at 5 and 10 years, respectively, while the global mortality rate is 31.8%. Of the seven deceased probands with MSMD, five were male and two were female. Meanwhile, five had IL-12Rβ1 deficiency (P1, P2, P6, P7, and P8), one had AR-complete IFN-gR1 deficiency (P17), and one had AD IFN-gR1 deficiency (P20). The median age at death was 4.5 years old (range: 3–16 years), and the only cause of death was multiorgan failure secondary to disseminated mycobacterial infection caused by BCG in six probands (P1, P2, P6, P7, P8, and P17) and an unidentified mycobacterial species in one (P20). Two patients had a mycobacterial disseminated infection and additionally in blood cultures grew Candida spp. (P2) and K. pneumoniae (P7).
Discussion
To the best of our knowledge, this is the first report of a Mexican cohort with MSMD that included 22 patients. In 2019, the number of registered births in Mexico reached two million, and the crude birth rate was 16.8 births per 1000 inhabitants nationwide in 2020 [32]. As MSMD affects approximately 1/50,000 individuals worldwide [2], the number of reported cases (confirmed at the genetic level) in Mexico suggests that MSMD is underdiagnosed. Other IEI, such as chronic granulomatous disease (CGD) or dysgammaglobulinemia, are more commonly diagnosed because of significant suspicion among physicians and central accessibility to screening tests [17, 18]. Among the 19 causal genes of MSMD [4], this study described variant in three genes (IL12RB1, IFNGR1, and IFNGR2) that define four genetic disorders (AR IL-12Rβ1 deficiency, AR-complete IFN-gR1 deficiency, AD-partial IFN-gR1 deficiency, and AR-partial IFN-gR2 deficiency). One of the most frequent variants in IL12RB1 Mexican kindreds is c.1791 + 2 T > G, which is the most frequently reported mutation worldwide [21, 33]. P10 had a compound heterozygous variant of IL12RB1, c.635G > A, that was only recently reported (21) and c.1750C > T, which has not been previously reported. This patient had null expression of IL-12Rβ1 on the surface of PHA-activated T cells; her parents were heterozygous carriers of each variant. In contrast, in IFNGR1, the most significant number of mutated alleles was found in exon 6 due to a deletion hotspot (c.819_822del), which often arises de novo [34]. One of the three described patients in this series harbored this mutation.
Similar to other countries, BCG vaccination is mandatory in Mexico [3, 35,36,37,38]. In this study, M. bovis-BCG was the main infectious agent in patients with MSMD, resulting in BCG being the principal cause of morbidity and mortality. In IL-12Rβ1 deficiency, patients who died secondary to BCG were diagnosed with an advanced stage of the infectious disease. Health policies in countries where BCG is mandatory must include warning signs and/or screening for MSMD in children with an adverse reaction to the BCG vaccine. Patients deficient in IL-12Rβ1 are also susceptible to Salmonella infections, which are associated with recurrent leukocytoclastic vasculitis [12]. One patient in this study developed nephritis with recurrent leukocytoclastic vasculitis. However, the link between the autoimmune manifestations of IL-12Rβ1 deficiency and Salmonella infections remains unclear [12, 39]. Interestingly, subcutaneous gammaglobulin ceased the relapses of Salmonella sepsis in P12, suggesting that subcutaneous gammaglobulin might be a therapeutic option in cases of recurrent Salmonella infection. Meanwhile, P13 presented with Salmonella brain abscess, which has been described in CGD patients [40, 41]. However, to our knowledge, this clinical manifestation has not yet been described in patients with MSMD. Infectious diseases, such as histoplasmosis and leishmaniasis, are endemic in Mexico [42, 43]. A few of our patients with IL-12Rβ1 deficiency developed histoplasmosis. Hence, MSMD should be suspected even in patients with histoplasmosis and without other more typical MSMD-related infections. Interestingly, leishmaniasis has been reported in IL-12Rβ1-deficient patients, yet we did not find leishmaniasis in our cohort [36, 44,45,46]. The mortality rate in the present study was 31.8%, which raises the question of whether these patients should undergo hematopoietic stem cell transplantation [47,48,49]
In conclusion, the clinical, immunological, and molecular characterization of this patient series with MSMD in Mexico confirmed data in previous studies. The p.S584P variant in IL12RB1 is first described. Because BCG vaccination is mandatory in Mexico, BCG infection was an important cause of morbidity and mortality; cases with adverse reactions to BCG must be investigated to rule out MSMD. To prevent life-threatening complications of BCG infection, BCG vaccination should be contraindicated in newborns with siblings and relatives with a history of MSMD. Patients with unexplained infections caused by endemic intracellular pathogens, such as histoplasma or other deep mycoses, should be suspected of having MSMD and studied for genetic defects in IFN-γ-mediated immunity. The detection of the responsible gene of MSMD will help, on the one hand, to define the treatment and prognosis, and on the other hand, to be able to make an understanding between clinical and genetic correlation.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
DGE. Manual de Procedimientos Estandarizados para la Vigilancia Epidemiológica de las Micobacteriosis. 2019. [Available from: https://epidemiologia.salud.gob.mx/gobmx/salud/documentos/manuales/18_Manual_Micobacteriosis.pdf.
Boisson-Dupuis S, Bustamante J. Mycobacterial diseases in patients with inborn errors of immunity. Curr Opin Immunol. 2021;72:262–71.
Azarsiz E, Karaca N, Karaca E, Aksu G, Genel F, Gulez N, et al. Eight years of follow-up experience in children with mendelian susceptibility to mycobacterial disease and review of the literature. Asian Pac J Allergy Immunol. 2021.
Bustamante J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. 2020;139(6–7):993–1000.
Pedraza-Sanchez S, Herrera-Barrios MT, Aldana-Vergara R, Neumann-Ordonez M, Gonzalez-Hernandez Y, Sada-Diaz E, et al. Bacille Calmette-Guerin infection and disease with fatal outcome associated with a point mutation in the interleukin-12/interleukin-23 receptor beta-1 chain in two Mexican families. Int J Infect Dis. 2010;14(Suppl 3):e256–60.
Pedraza S, Lezana JL, Samarina A, Aldana R, Herrera MT, Boisson-Dupuis S, et al. Clinical disease caused by Klebsiella in 2 unrelated patients with interleukin 12 receptor beta1 deficiency. Pediatrics. 2010;126(4):e971–6.
de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 2010;89(6):381–402.
Martinez-Morales MC, Deswarte C, Castaneda-Casimiro J, Bustamante J, Blancas-Galicia L, Scheffler-Mendoza S. Disseminated infection by M. tuberculosis complex in patient with IFN-gamma receptor 1 complete deficiency. Rev Alerg Mex. 2017;64(4):499–504.
Leon-Lara X, Hernandez-Nieto L, Zamora CV, Rodriguez-D’Cid R, Gutierrez MEC, Espinosa-Padilla S, et al. Disseminated infectious disease caused by histoplasma capsulatum in an adult patient as first manifestation of inherited IL-12Rbeta1 deficiency. J Clin Immunol. 2020;40(7):1051–4.
Ramirez-Alejo N, Blancas-Galicia L, Yamazaki-Nakashimada M, Garcia-Rodriguez SE, Rivas-Larrauri F, Paolo-Cienfuegos DP, et al. Molecular analysis for patients with IL-12 receptor beta1 deficiency. Clin Genet. 2014;86(2):161–6.
Moncada-Velez M, Martinez-Barricarte R, Bogunovic D, Kong XF, Blancas-Galicia L, Tirpan C, et al. Partial IFN-gammaR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. 2013;122(14):2390–401.
Blancas-Galicia L, Penafiel-Vicuna AK, Scheffler-Mendoza S, Rojas-Maruri M, Rivas-Larrauri F, Rodriguez-Lozano AL, et al. Recurrent Salmonella infections and nephritis complicating IgA vasculitis in a patient with IL12-RB1 deficiency. J Investig Allergol Clin Immunol. 2021:0.
Allen-Manzur JG, Espinosa-Padilla SE, Bustamante J, Blancas-Galicia L, Mendieta-Flores E. Disseminated infection caused by the bacillus Calmette-Guerin vaccine and SARS-CoV-2 coinfection in a patient with IL-12 receptor beta1 subunit deficiency. Rev Alerg Mex. 2020;67(4):401–7.
Staines-Boone AT, Deswarte C, Venegas Montoya E, Sanchez-Sanchez LM, Garcia Campos JA, Muniz-Ronquillo T, et al. Multifocal recurrent osteomyelitis and hemophagocytic lymphohistiocytosis in a boy with partial dominant IFN-gammaR1 deficiency: case report and review of the literature. Front Pediatr. 2017;5:75.
Rosain J, Oleaga-Quintas C, Deswarte C, Verdin H, Marot S, Syridou G, et al. A variety of Alu-mediated copy number variations can underlie IL-12Rbeta1 deficiency. J Clin Immunol. 2018;38(5):617–27.
Pedraza-Sanchez S, Mendez-Leon JI, Gonzalez Y, Ventura-Ayala ML, Herrera MT, Lezana-Fernandez JL, et al. Oral administration of human polyvalent IgG by mouthwash as an adjunctive treatment of chronic oral candidiasis. Front Immunol. 2018;9:2956.
Garcia-Dominguez M, Valero-Galvez GC, Velazquez-Rios CA, Blancas-Galicia L. Registry of inborn errors of immunity in a pediatric hospital. Rev Alerg Mex. 2020;67(3):268–78.
Blancas-Galicia L, Santos-Chavez E, Deswarte C, Mignac Q, Medina-Vera I, Leon-Lara X, et al. Genetic, immunological, and clinical features of the first Mexican cohort of patients with chronic granulomatous disease. J Clin Immunol. 2020;40(3):475–93.
Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E, Jr, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138(1):241–8 e3.
Regiones de México. [Available from: https://es.wikipedia.org/wiki/Regiones_de_M%C3%A9xico.
van de Vosse E, Haverkamp MH, Ramirez-Alejo N, Martinez-Gallo M, Blancas-Galicia L, Metin A, et al. IL-12Rbeta1 deficiency: mutation update and description of the IL12RB1 variation database. Hum Mutat. 2013;34(10):1329–39.
Zhou X, Jia W, Ni Z, Wang A, Liu Z, Hou M, et al. Three novel compound heterozygous IL12RB1 mutations in Chinese patients with Mendelian susceptibility to mycobacterial disease. PLoS ONE. 2019;14(4): e0215648.
PopViz. [Available from: http://shiva.rockefeller.edu/PopViz.
Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26(6):454–70.
Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, et al. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine (Baltimore). 2013;92(2):109–22.
Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, et al. Partial recessive IFN-gammaR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20(8):1509–23.
Gunn JS, Marshall JM, Baker S, Dongol S, Charles RC, Ryan ET. Salmonella chronic carriage: epidemiology, diagnosis, and gallbladder persistence. Trends Microbiol. 2014;22(11):648–55.
Siberil S, Elluru S, Graff-Dubois S, Negi VS, Delignat S, Mouthon L, et al. Intravenous immunoglobulins in autoimmune and inflammatory diseases: a mechanistic perspective. Ann N Y Acad Sci. 2007;1110:497–506.
Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18(6):347–61.
Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–21.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
[Available from: https://es.statista.com/.
Sarrafzadeh SA, Nourizadeh M, Mahloojirad M, Fazlollahi MR, Shokouhi Shoormasti R, Badalzadeh M, et al. Molecular, immunological, and clinical features of 16 Iranian patients with mendelian susceptibility to mycobacterial disease. J Clin Immunol. 2019;39(3):287–97.
van de Vosse E, van Dissel JT. IFN-gammaR1 defects: mutation update and description of the IFNGR1 variation database. Hum Mutat. 2017;38(10):1286–96.
Ying W, Liu D, Dong X, Wang W, Hui X, Hou J, et al. Current status of the management of Mendelian susceptibility to mycobacterial disease in Mainland China. J Clin Immunol. 2019;39(6):600–10.
Taur PD, Gowri V, Pandrowala AA, Iyengar VV, Chougule A, Golwala Z, et al. Clinical and molecular findings in Mendelian susceptibility to mycobacterial diseases: experience from India. Front Immunol. 2021;12:631298.
Mahdaviani SA, Mansouri D, Jamee M, Zaki-Dizaji M, Aghdam KR, Mortaz E, et al. Mendelian Susceptibility to Mycobacterial Disease (MSMD): clinical and genetic features of 32 Iranian patients. J Clin Immunol. 2020;40(6):872–82.
Indumathi CK, Bustamante J. Clinical and immunological profile of children with Mendelian Susceptibility to Mycobacterial Diseases (MSMD) from an Indian tertiary care hospital. Indian J Tuberc. 2021;68(2):292–7.
Gokturk B, Reisli I, Caliskan U, Oleaga-Quintas C, Deswarte C, Turul-Ozgur T, et al. Infectious diseases, autoimmunity and midline defect in a patient with a novel bi-allelic mutation in IL12RB1 gene. Turk J Pediatr. 2016;58(3):331–6.
Finocchi A, Claps A, Serafinelli J, Salfa I, Longo D, Di Matteo G, et al. Chronic granulomatous disease presenting with salmonella brain abscesses. Pediatr Infect Dis J. 2014;33(5):525–8.
Sarria JC, Vidal AM, Kimbrough RC 3rd. Salmonella enteritidis brain abscess: case report and review. Clin Neurol Neurosurg. 2000;102(4):236–9.
Laniado-Laborin R. Coccidioidomycosis and other endemic mycoses in Mexico. Rev Iberoam Micol. 2007;24(4):249–58.
Salud Psd. Prevención y control de Leishmaniasis 2018 [Available from: http://www.cenaprece.salud.gob.mx/descargas/pdf/PAE_PrevencionControlLeishmaniasis2013_2018.pdf.
Parvaneh N, Barlogis V, Alborzi A, Deswarte C, Boisson-Dupuis S, Migaud M, et al. Visceral leishmaniasis in two patients with IL-12p40 and IL-12Rbeta1 deficiencies. Pediatr Blood Cancer. 2017;64(6).
Sanal O, Turkkani G, Gumruk F, Yel L, Secmeer G, Tezcan I, et al. A case of interleukin-12 receptor beta-1 deficiency with recurrent leishmaniasis. Pediatr Infect Dis J. 2007;26(4):366–8.
Tan C, Cagdas-Ayvaz D, Metin A, Keskin O, Tezcan I, Sanal O. Clinical and genetic features of IL12Rb1 deficiency: single center experience of 18 patients. Turk J Pediatr. 2016;58(4):356–61.
Nazir HF, Rawas AA, Tamemi SA, Zadjali SA, Hosni SA, Tauro M, et al. Hematopoietic stem cell transplantation for patients with autosomal recessive complete INF-lambda receptor 2 deficiency: experience in Oman. Transplant Cell Ther. 2021;27(10):881 e1- e5.
Patel S, Uppuluri R, Vellaichamy Swaminathan V, Ravichandran N, Melarcode Ramanan K, Raj R. Mendelian susceptibility to mycobacterial disease-challenges in hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2020;67(5):e28187.
Radwan N, Nademi Z, Lum SH, Flood T, Abinun M, Owens S, et al. Outcome of hematopoietic stem cell transplantation in patients with Mendelian susceptibility to mycobacterial diseases. J Clin Immunol. 2021;41(8):1774–80.
Acknowledgements
We would like to thank the patients, their relatives, and physicians. We also thank Yelena Nemirovskaya, Dana Liu, Christine Rivalain, Maya, Chrabieh, and Lazaro Lorenzo-Diaz for their administrative support.
Funding
M.A.Y.N., J.C.B.O., S.L.R, S.E.P., and L.B.G. are SNI-CONACYT members. The UIID was funded in part by FUMENI A.C. The Laboratory of Human Genetics of Infectious Diseases was funded in part by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, Institut National de la Santé et de la Recherche Médicale (INSERM), University of Paris, National Institutes of Health (NIH) (R01AI088364, R01AI095983, R01AI127564, R01AI163029, UL1TR001866), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency (ANR) under the “Investments for the future” program (grant number ANR-10-IAHU-01), ANR-GENMSMD/ANR-16-CE17-0005–01 (for J. B.), ECOS-NORD (C19S01-63407), the ANRS Nord-Sud (ANRS-COV05), the French Foundation for Medical Research (FRM) (EQU201903007798), ANR FNS LTh-MSMD-CMCD (ANR-18-CE93-0008–01), ANR GENVIR (ANR-20-CE93-003), ANR AABIFNCOV (ANR-20-CO11-0001), the European Union’s Horizon 2020 research and innovation program under grant agreement No 824110 (EASI-genomics), REACTing-INSERM, the Square Foundation, Grandir—Fonds de solidarité pour l’enfance, the SCOR Corporate Foundation for Science, and the French Ministry of Higher Education, Research, and Innovation (MESRI-COVID-19). Institut National de la Santé et de la Recherche Médicale (INSERM), REACTing-INSERM, and the University Paris Cité.
Author information
Authors and Affiliations
Contributions
A.K.P.V., K.X.L., S.P.S., A.L.I., Y.G., M.T., E.M.T., and C.S. performed the experiments. M.A.Y.N., E.M.F., M.E.N.N., J.C.L.R., L.H.N., M.G.R.M., J.B.S., J.L.L.H., C.M.R.M., J.C.B.O, O.Z.M., T.S.B., E.V.M, N.E.A.G., S.L.R., A.P., and E.G.S. diagnosed and treated the patients. E.J. and S.B.D. performed the genetic analysis. L.B.G. and K.X.L.L. drafted the manuscript. L.B.G., S.P.S., J.L.C., F.E.R., S.E.P., and J.B. supervised this study. All the authors have discussed, revised, and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The experiments described here were performed in Mexico and France in accordance with local regulations and with the approval of the Institutional Review Board of Necker Hospital for Sick Children, France.
Consent to Participate
Written informed consent to participate was obtained from the parents of the patient. Informed consent for participation in this study was obtained in accordance with local regulations, with approval from the relevant Institutional Review Boards.
Consent for Publication
Consent for publication was obtained from the patient’s parents. All authors approved the final version of the manuscript.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Peñafiel Vicuña, A.K., Yamazaki Nakashimada, M., León Lara, X. et al. Mendelian Susceptibility to Mycobacterial Disease: Retrospective Clinical and Genetic Study in Mexico. J Clin Immunol 43, 123–135 (2023). https://doi.org/10.1007/s10875-022-01357-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01357-8