
Abstract
Quantum mechanical calculations are presented that predict that one-bond deuterium isotope effects on the 15N chemical shift of backbone amides of proteins, 1Δ15N(D), are sensitive to backbone conformation and hydrogen bonding. A quantitative empirical model for 1Δ15N(D) including the backbone dihedral angles, Φ and Ψ, and the hydrogen bonding geometry is presented for glycine and amino acid residues with aliphatic side chains. The effect of hydrogen bonding is rationalized in part as an electric-field effect on the first derivative of the nuclear shielding with respect to N–H bond length. Another contributing factor is the effect of increased anharmonicity of the N–H stretching vibrational state upon hydrogen bonding, which results in an altered N–H/N–D equilibrium bond length ratio. The N–H stretching anharmonicity contribution falls off with the cosine of the N–H···O bond angle. For residues with uncharged side chains a very good prediction of isotope effects can be made. Thus, for proteins with known secondary structures, 1Δ15N(D) can provide insights into hydrogen bonding geometries.
Similar content being viewed by others

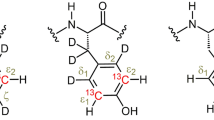

Introduction
Deuterium isotope effects on chemical shifts have proved to be a sensitive gauge for hydrogen bonding (Jameson 1991; Abildgaard et al. 1998; Dziembowska et al. 2004; Kim et al. 2006). Deuterium substitution at the N–H hydrogen site leads to one-bond isotope effects on the 15N chemical shift: 1Δ15N(D) = σ15N(D)−σ15N(H) = δ15N(H)−δ15N(D). This “difference” is caused by a small change in the (1) vibrational state due to the altered reduced mass upon deuteriation, and (2) equilibrium geometry due to anharmonicity of the N–H stretching mode potential energy surface. Deuterium substitution is favorable for the study of isotope effects due to the large relative change in mass. 15N is a good nucleus for observation because of its large chemical shift range. 1Δ15N(D) reports on the hydrogen-bonding geometry in ammonium ions (Munch et al. 1992), and Jaravine et al. (2004) found that 1Δ15N(D) values in ubiquitin could be expressed as a linear function of the 15N chemical shift and the trans-hydrogen bond scalar coupling h3 J NC′. One-bond deuterium isotope effects on 13Cα (1Δ13Cα(D)) have been shown to correlate with protein backbone conformation (LeMaster et al. 1994) and in principle can be used like 1Hα, 13Cα, and 13Cβ chemical shifts and 1 J CαHα for distinguishing α-helix and β-strand secondary structures (Wishart and Case 2001). Our objective here is to demonstrate how 1Δ15N(D) is determined by protein backbone structure, and in addition by the hydrogen-bonding geometry (Fig. 1).

The N-formyl or N-acetylaminoacidamides used in the calculations. All heavy atom torsion angles, the R N–O distance, the θ C′–N···O angle and the ϕC′–N···O−C′ dihedral angle were kept at the experimental X-ray values. All other bond length and angles, and all hydrogen positions were geometry optimized at the BPW91/6-31G(d) or RHF/6-31G(d) level
Materials and methods
NMR measurements
Two-dimensional deuterium-decoupled HA(CACO)N experiments (Wang et al. 1995; Ottiger and Bax 1997) were recorded on a sample of commercially obtained human ubiquitin, uniformly enriched in 13C and 15N (VLI Research, PA, USA) and dissolved in a solution of [D2O]/[H2O] = 1.6, prepared in a manner similar to that described previously (LiWang and Bax 1996). Two spectra were collected as 512* (t 1) × 256* (t 2) data sets (where n* refers to n complex data points), with acquisition times of 389 ms (t 1) and 85 ms (t 2), where t 1 refers to the 15N dimension and t 2 to the 1H dimension. The total measuring time was 8 h for each experiment. Prior to Fourier transformation, the data were apodized with sine bell and squared sine bell functions shifted by 60° in both dimensions, and then zero-filled to 2,048* and 1,024* in the t 1 and t 2 dimensions, respectively. Data were processed using the program nmrPipe (Delaglio et al. 1995), and the peak positions and intensities for non-overlapping resonances were determined interactively using the program PIPP (Garrett et al. 1991; Fig. 2).

A small region of the 2D HA(CACO)N spectrum of human ubiquitin at pH 4.7 labeled with deuterium at the NH group ([D2O]/[H2O] = 1.6) recorded at 600 MHz 1H frequency, 25°C. For each backbone amide group, the upfield and downfield peaks originate from the deuterated and protonated isotopomers, respectively. The residue labels are according to the 15N chemical shifts
Molecular geometry
X-PLOR (Brünger 1987) calculations were performed on the X-ray crystal structure of ubiquitin [PDB ID: 1ubq] (Vijay-Kumar et al. 1987) using the “parhcsdx.pro” and “tophcsdx.pro” hydrogen parameter and topology files (Engh and Huber 1991), respectively, for adding and relaxing hydrogen positions. The water TIPS3P parameters were taken from param19.sol and toph19.sol (Jorgensen et al. 1983). This structure is in the following referred to as XPLOR.
Hydrogen bonding partners in human ubiquitin were identified between N–H and all possible hydrogen bond acceptors including crystallographic water oxygens. The hydrogen-bonds included all had an energy larger than 3 kcal for all types of donor–acceptor interactions, and the heavy atom equilibrium distance was less than 3 Å. The hydrogen bonding pattern was therefore established on a purely geometric basis. All shielding calculations of the individual amino acids of ubiquitin were performed on two types of RHF/6-31G(d) partially relaxed peptides: one on the formyl and amide end-capped amino acids as described below, and one with an additional formamide molecule in the hydrogen bonding partner position.
The structures of the individual amino acids used in the calculations were cloned from the protein X-ray structure including the preceding carbonyl and following amide groups. Hydrogen atoms were added to the resulting N-formyl-amino acid-amides and relaxed at the RHF/6-31G(d) level, and so were all heavy atom bond lengths and bond angles. Only the heavy atom dihedral angles were retained from the X-ray structure. The geometry of the structures of N-formyl-amino acid-amides with hydrogen bonding partners were obtained by supplementing the BPW91/6-31G(d) or RHF/6-31G(d) relaxed structures with a formamide molecule as hydrogen bond acceptor in a position corresponding to what is found in the crystal structure. This structure was relaxed as described above, but this time also keeping the N···Oformamide distance, the C′ i−1–N···Oformamide angle, and the C′ i−1–N···O = C′formamide dihedral angle at the X-ray value (Fig. 1 and Supplemental Table 1). These structures are referred to as BPW91 and RHF.
Model calculations on formamide and its dimer
Formamide and its hydrogen-bonded dimer were geometry optimized at the MP2 = FULL/6-311G(d,p) level using Gaussian94 (Frisch et al. 1995). Normal mode analysis and harmonic frequency calculations were performed at this level on the two structures. The potential energy scans of the N–H bond involved in the hydrogen bond (Fig. 3) were performed at the same level of theory but the geometry was that of identical monomers and the scanned hydrogen bond was straightened to 180° from the equilibrium value of 171° found both between the two identical monomers and the fully optimized dimer.

The calculated potential energy for the N–H bond displacement of monomeric and dimeric formamide. The bond lengths are given in Angstrom. The open and solid circles are the ab initio calculated values for the monomer and dimer, respectively. The curves are the fitted Morse functions to the calculated values. The zero point displacement values, ΔR N–H(D), are 0.0061 and 0.0071 Å for the monomer and dimer potentials, respectively, as calculated from the fitted Morse functions. The zero point energies and displacements are shown for the dimer potential. For ΔE < 50 kcal, the dimer potential can be described by ΔE = 106.95 {1−exp[−2.22 (R N–H−1.011352)]}2
Frequency calculations were done on a fully optimized dimer (Supplemental Table 2). Both the fully optimized monomer and the dimer yielded one and three negative frequencies in the normal mode calculation, respectively, indicating that the –NH2 group is not flat at the MP2 = FULL/6-311G(d,p) level of theory. The first-order energy gradients were zero for the flat structure which was at least a local stationary point, but a negative eigenvalue in the frequency calculation gave an inversion type out-of-plane motion involving the –NH2 atoms. We do not expect this to cause problems since we are only seeking a qualitative picture of the anharmonicity in the N–H stretching caused by hydrogen bonding, and so we chose to work with the planar dimer composed of identical monomers in a linear hydrogen bond.
A number of other considerations must be mentioned in this connection. The zero-point energy of the N–H stretching mode was calculated within the harmonic approximation, and is thus not exact. We did not scan the entire normal mode motion but only the N–H displacement, keeping the remaining atomic positions fixed. In the frequency calculation of the dimer, the normal modes of the transitions at 3,654 and 3,801 cm−1 are strongly coupled, representing symmetric and asymmetric N–H2 stretching, respectively. This coupling is entirely lost when substituting the protein relevant N–H hydrogen with deuterium.
Results and discussion
In the present work we test a simple method for the calculation of 1Δ15N(D) isotope effects by simple uniform geometric perturbations, and compare the calculated values with experimental results collected on the protein human ubiquitin. We also present an empirical correlation between the experimental 1Δ15N(D) data and predicted values based on secondary structure and hydrogen bonding.
A small section of the deuterium-decoupled HA(CACO)N spectrum is shown in Fig. 2. Each 1Hα shows two cross peaks to the 15N resonance of the next residue, corresponding to the protonated and deuterated states of the amide. The excellent F 1 resolution and the high signal-to-noise ratio make it possible to measure the 1Δ15N(D) isotope effects with very high precision (±0.001 ppm). Isotope effects were measured from the relative displacement in the F 1 dimension caused by deuteriation, using the correlation between 1H α i and N i+1(H) and N i+1(D), respectively.
Shown in Fig. 4 is a plot of experimental 1Δ15N(D) versus δ15N. There seems to be a linear correlation between 1Δ15N(D) and δ15N in the non-hydrogen bonded N–H groups. This correlation, which is lost with hydrogen bonding, suggests that the intrinsic hydrogen–deuterium isotope effect is proportional to the heavy atom chemical shift, which has also been observed by Jaravine et al. (2004). 1Δ15N(D) values seem to group with secondary structure and hydrogen bonding with better separation in 1Δ15N(D) dimension than in the δ1HN or δ15N dimensions. Notice the small spread in the non-hydrogen bonded β-sheet values compared to the hydrogen bonded ones, both in 1Δ15N(D) and δ1HN (Supplemental Fig. 1), indicating a large effect of hydrogen bonding on these parameters. Clearly the separation between β-sheet and α-helix 1Δ15N(D) values is not as good as that observed for the chemical shift and coupling constants of the 13Cα group [δ1Hα, δ13Cα and 1 J CαHα] (Spera and Bax 1991; Wishart et al. 1991; Vuister et al. 1992; Wishart et al. 1992), but the 1Δ15N(D) data seems to include hydrogen bonding information.

Experimental 1Δ15N(D) values plotted against experimental 15N chemical shifts, δ15N, from human ubiquitin. The correlation line, 1Δ15N(D) = 0.0054 δ15N + 0.0754 ppm (R 2 = 0.93), is for non-hydrogen bonded N–H groups. The 15N chemical shift data are not random coil corrected or corrected for neighboring residue offset
1Δ15N(D) can be approximated by the following equation (Jameson 1991):
where σ is the NMR shielding constant of 15N in ppm, dσ/dR N–H is the first derivative of the shielding constant with respect to the amide N–H bond length in ppm/Å, and ΔR N–H(D) = R N–H−R N–D is the difference in the mean bond lengths of N–H and N–D in Angstrom (Jameson 1991; Abildgaard et al. 1998; Dziembowska et al. 2004; Kim et al. 2006).
We have calculated the shape of the N–H potential energy surface with and without a hydrogen bonding partner (Fig. 3). The N–H potential energy surfaces from the monomer and the dimer can be reproduced by Morse functions. In the non-hydrogen bonded case, we get a difference in equilibrium N–H bond lengths of ΔR N–H(D) = 0.0061 Å. The increase in ΔR N–H(D) upon hydrogen bonding is 0.001 Å, which is an increase of 16 and 22% using the Morse (0.0061 to 0.0071 Å) and ab initio (0.0050 to 0.0061 Å) frequencies, respectively (Supplemental Table 2). Using a value of 100 ppm/Å (this value is the approximate calculated slope of the shielding surface, see Supplemental Table 3) for the first derivative of the shielding surface with respect to N–H bond length the estimated increase in 1Δ15N(D) upon hydrogen bonding is ~0.1 ppm, which is ~70% of the entire variation of the experimentally observed 1Δ15N(D) (see Table 2). The calculations on the idealized formamide dimer probably represent a maximum with respect to equilibrium bond length displacement with deuterium substitution, as the N···O distance is shorter in the amide dimer than in the protein. This contribution from the increased anharmonicity of the N–H stretching should be a positive contribution when subjecting the amide to hydrogen bonding.
Using Eq. 1 1Δ15N(D) can be calculated provided the first derivative of the shielding constant (see Supplemental Table 3) and the change in the NH bond length upon deuteriation are known. In order to get a feeling for the importance of the various factors we have done the following. Calculations using a bond length change of 0.0061 Å (see above) and amino acid geometries obtained from the X-ray crystal structure but neglecting hydrogen bonding show a poor correlation between calculated and experimental 1Δ15N(D) values (blue bars, Fig. 5). Including formamide as hydrogen-bond acceptor and the Morse calculated value of ΔR N–H(D) = 0.0071 Å for the hydrogen bonded amino acids the calculated values are in slightly better agreement with experimental results (black bars, Fig. 5). However, it is obvious that none of the calculated values are very good as a standard bond extension has been used. Obviously, for each hydrogen bonded pair a value has to be calculated. This is not very practical and very time consuming. We have therefore taken different approach, see below.

Calculated and experimental 1Δ15N(D) values for aliphatic and glycine residues of ubiquitin. Experimental values are shown in yellow and were collected using the 2H-decoupled HA(CACO)N experiment described earlier (Wang et al. 1995) on a sample of human ubiquitin equilibrated in a [D2O]/[H2O] = 1.6 solvent mixture, pH 4.7, 25°C. QM calculations using N-formylaminoacid-amides (Fig. 1) without hydrogen bond partners are shown in blue. Inclusion of electric-field effects of hydrogen bonding and the Morse calculated value of 0.0071 Å for ΔRN–H(D) yields the calculated 1Δ15N(D) values shown in black
(For other calculated isotope effects using Eq. 1 see Supplemental Table 3).
As shown in Table 1, calculated 1Δ15N(D) for N-formylglycineamide (Fig. 1) depends on backbone dihedral angles, with a Φ/Ψ combination of 180°/180° giving a value of 0.79 ppm for the non-hydrogen bonded case (marked with ∞), which is slightly smaller compared to 0.94, 0.94 and 0.97 ppm for 180°/90°, 90°/180°, and 90°/90°, respectively. QM calculations were also used to estimate electric-field effects from hydrogen bonding on the shielding derivative, dσ/dR N–H. Introduction of a water molecule as a hydrogen-bond acceptor for the N i –H hydrogen (Fig. 6) has the effect of decreasing the shielding derivative (see Eq. 1), thereby decreasing 1Δ15N(D). The distance dependence of 1Δ15N(D) on the proximity of the water molecule is smooth as seen from Table 1. For a linear hydrogen bond with an NH···OH2 distance of 1.8 Å and Φ/Ψ = 180°/180°, 1Δ15N(D) decreases by 0.26 ppm, whereas decreases of ~0.52 ppm are found for other Φ/Ψ combinations. The difference is probably due to electric fields in the N i –H bond direction, which are largest for the 180°/180° conformation because the carbonyl oxygen of C′ i lies close to and at the smallest angle to the N–H bond (Hansen et al. 1994) (dashed green line in Fig. 5). In calculations of 1Δ15N(D) of ammonium ions surrounded by water, it was found that a negative charge could mimic a water molecule to a good extent (Munch et al. 1992), which bears witness to the electric-field nature of this effect.

Drawing of a water molecule hydrogen-bonded to the N–H group of N-formylglycineamide. White, grey, blue, and red colors are used for hydrogen, carbon, nitrogen, and oxygen atoms, respectively. The dashed green line is along the C i–1′N i bond. Backbone torsion angles Φ and Ψ are shown in the figure
Thus, calculations suggest that 1Δ15N(D) are sensitive to (1) backbone conformation, (2) electric-field effects and (3) anharmonicity effects from hydrogen bonding. We therefore performed a fit of the experimental 1Δ15N(D) data against backbone dihedral angles and geometric parameters of the hydrogen bonds for all amino acids with aliphatic side chains and glycine (Table 2) and obtained the following empirical equation for 1Δ15N(D):
where Φ′ = cos(Φ + 88° ± 8°), Ψ′ = cos(Ψ−68° ± 9°), A = 0.66 ± 0.02 ppm, B = 0.06 ± 0.01 ppm, C = 0.043 ± 0.009 ppm Å, and D = 0.03 ± 0.02 ppm. The coefficients are the averages of 10,000 fits where each time six out of 22 data points were randomly removed, and the values of the coefficients A, B, and C were randomly set between −1.0 and +1.0 ppm prior to each fit, whereas D was randomly set between −5.0 and +5.0 ppm. The standard deviations of the averages from these fits are used as estimates of the uncertainties in the coefficients. Angles were obtained as explained above using the BPW91/6-31G(d) level of theory. F-test analysis showed that including the factor 1/r, where r is the hydrogen bond distance, in term D does not statistically improve the fit for Eq. 2. A plot of the experimental 1Δ15N(D) values versus predictions from Eq. 2 for the protein ubiquitin are shown in Fig. 7. The pairwise root mean square difference between the experimental and predicted 1Δ15N(D) values is 0.014 ppm.

Experimental versus predicted 1Δ15N(D) values for aliphatic residues using Eq. 2 for the protein ubiquitin. The solid circle and solid squares are for (apparently) non-hydrogen bonded backbone amides of glycine and leucine residues, respectively. The line is along the diagonal. The residues used in this plot are listed in Table 2. The pairwise root mean square difference between the experimental and calculated values is 0.014 ppm
The Φ′ and Ψ′ terms show that there is a large variation with backbone conformation, which is consistent with the dependence of 1Δ15N(D) on the chemical shift of 15N reported by Jaravine et al. (2004). The third term contains both electric field and anharmonicity effects of hydrogen bonding. The cosine contribution to the third term indicates that the hydrogen bonding effect falls off with bending of the hydrogen bond. It should be noted that although Eq. 2 does not have a solid theoretical or physical basis, it reveals that secondary structure and hydrogen bonding strongly influence 1Δ15N(D).
In Supplemental Fig. 2a, 1Δ15N(D) of the remaining non-aliphatic amino acids are predicted with the aliphatic parameters and using RHF/6-31G(d) optimized geometries. The non-aliphatic amino acids lack accurate estimates of the electric fields generated by the polar side chains and can be flexible and less well determined by X-ray and NMR methods. For these residues Eq. 2 produces a poorer correlation with experimental 1Δ15N(D) values.
The 1Δ15N(D) isotope effect can be determined with great precision in proteins, and predicted for amino acids with uncharged side-chains provided that (1) relaxed hydrogen bond geometries are used, (2) hydrogen bonding partners are included, (3) the shortening of the N–H bond length upon deuteriation is applied, and (4) charges from neighboring atoms are taken into account in a more elaborate electric-field treatment.
Theoretical calculations are possible using even very low basis sets: RHF/6-31G(d) and BPW91/6-31G(d,p) relaxed hydrogen bond geometries gave similar results (Supplemental Fig. 2b). The experimental values can be reproduced by an equation involving the backbone angles, the change in the bond length upon deuteriation, and hydrogen bonding geometries. One-bond isotope effects are a promising tool in optimizing hydrogen-bond geometries in proteins, provided the secondary structures are known. Recently, deuterium isotope effects measured on side chain amides of asparagines and glutamine residues were shown to be sensitive to hydrogen bonding and charge effects, and can be used as a double check on the rotamer conformations of these residues (Liu et al. 2008).
References
Abildgaard J, Bolvig S, Hansen PE (1998) Unraveling the electronic and vibrational contributions to deuterium isotope effects on 13C chemical shifts using ab initio model calculations. Analysis of the observed isotope effects on sterically perturbed intramolecular hydrogen-bonded o-hydroxy acyl aromatics. J Am Chem Soc 120:9063–9069
Brünger AT (1987) X-PLOR version 3.1 a system for X-ray crystallography and NMR. Yale University Press, New Haven
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Dziembowska T, Hansen PE, Rozwadowski Z (2004) Studies based on deuterium isotope effect on 13C chemical shifts. Prog Nucl Magn Reson Spectrosc 45:1–29
Engh RA, Huber R (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Cryst A47:392–400
Frisch MJ, Trucks GW, Schlegel HB, Gill PMW, Johnson BG, Robb MA, Cheeseman JR, Keith TA, Petersson GA, Montgomery JA, Raghavachari K, Al-Laham MA, Zakrzewski VG, Ortiz JV, Foresman JB, Cioslowski J, Stefanov BB, Nanayakkara A, Challacombe M, Peng CY, Ayala PY, Chen W, Wong MW, Andres JL, Replogle ES, Gomperts R, Martin RL, Fox DJ, Binkley JS, Defrees DJ, Baker J, Stewart JP, Head-Gordon M, Gonzalez C, Pople JA (1995) Gaussian 95, revision A.1. Gaussian Inc., Pittsburgh
Garrett DS, Powers R, Gronenborn AM, Clore GM (1991) A common sense approach to peak peaking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Res 95:214–220
Hansen PE, Abildgaard J, Hansen AE (1994) Ab initio calculations of external charge effects on the isotropic 13C, 15N and 17O nuclear shieldings of amides. Chem Phys Lett 224:275–282
Jameson CJ (1991) The dynamic and electronic factors in isotope effects on NMR parameters. In: Buncel E, Jones JR (eds) Isotopes in the physical and biomedical sciences, vol 2. Elsevier, Amsterdam, pp 1–54
Jaravine VA, Cordier F, Grzesiek S (2004) Quantification of H/D isotope effects on protein hydrogen-bonds by h3JNC′ and 1JNC′ couplings and peptide group 15N and 13C′ chemical shifts. J Biomol NMR 29:309–318
Jorgensen WL, Chandrasekhar J, Madura JD (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Kim Y-I, Manalo MN, Pérez LM, LiWang A (2006) Computational and empirical trans-hydrogen bond deuterium isotope shifts suggest that N1–N3 A:U hydrogen bonds of RNA are shorter than those of A:T hydrogen bonds of DNA. J Biomol NMR 34:229–236
LeMaster DM, LaIuppa JC, Kushlan DM (1994) Differential deuterium isotope shifts and one-bond 1H–13C scalar couplings in the conformational analysis of protein glycine residues. J Biomol NMR 4:863–870
Liu A, Wang J, Lu Z, Yao L, Li Y, Yan H (2008) Hydrogen-bond detection, configuration assignment and rotamer correction of side-chain amides in large proteins by NMR spectroscopy through protium/deuterium isotope effects. Chembiochem 9:2860–2871
LiWang A, Bax A (1996) Equilibrium protium/deuterium fractionation of backbone amides in U-13C/15N labeled human ubiquitin by triple resonance NMR. J Am Chem Soc 118:12864–12865
Munch M, Hansen AE, Hansen PE, Bouman TD (1992) Ab initio calculations of deuterium isotope effects on hydrogen and nitrogen nuclear magnetic shielding in the hydrated ammonium ion. Acta Chem Scand 46:1065–1071
Ottiger M, Bax A (1997) An empirical correlation between amide deuterium isotope effects on 13Cα chemical shifts and protein backbone conformation. J Am Chem Soc 119:8070–8075
Spera S, Bax A (1991) Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J Am Chem Soc 113:5490–5492
Vijay-Kumar S, Bugg CE, Cook WJ (1987) Structure of ubiquitin refined at 1.8 Å resolution. J Mol Biol 194:531–544
Vuister GW, Delaglio F, Bax A (1992) An empirical correlation between 1 J CαHα and protein backbone conformation. J Am Chem Soc 114:9674–9675
Wang AC, Grzesiek S, Tschudin R, Lodi PJ, Bax A (1995) Sequential backbone assignment of isotopically enriched proteins in D2O by deuterium-decoupled HA(CA)N and HA(CACO)N. J Biomol NMR 5:376–382
Wishart DS, Case DA (2001) Use of chemical shifts in macromolecular structure determination. Methods Enzymol 338:3–34
Wishart DS, Sykes BD, Richards FM (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol 222:311–333
Wishart DS, Sykes BD, Richards FM (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31:1647–1651
Acknowledgments
A.L. is grateful to UC Merced for funding. We are also grateful to Professor Aage E. Hansen and Dr. Ad Bax for advice, and Stefanie Wengrzik for help with data analysis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Abildgaard, J., Hansen, P.E., Manalo, M.N. et al. Deuterium isotope effects on 15N backbone chemical shifts in proteins. J Biomol NMR 44, 119–126 (2009). https://doi.org/10.1007/s10858-009-9316-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-009-9316-0