
Abstract
In the present study we investigate the structural, Fermi surface and optical properties of the recently synthesized (Zr3−x Ti x )AlC2 MAX phases using density functional theory. The inclusion of Ti in the M site causes a reduction of both the a and c lattice parameters. The reduction of the a lattice parameter with respect to Ti content is more significant and this is reflected in the increase of the c/a ratio. The Fermi surfaces are formed mainly due to the low-dispersive transition metal d-like orbitals, which are responsible for the conductivity in the compounds. The energy loss spectra reveals that the two end members of (Zr3−x Ti x )AlC2 (x = 0 and 3) change from metallic to dielectric response at incident light energies 11.9, 13.4 and 10.8, 13.5 eV for [100] and [001] polarization directions, respectively. The reflectivity spectra indicate the ability of two end compounds Zr3AlC2 and Ti3AlC2 to be candidate materials for coatings to reduce solar heating. The calculated optical functions show the dependence on the polarization directions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
MAX phases are a class of solids, which have a unique combination of outstanding physical and chemical properties. These metallic and ceramic properties are due to the highly anisotropic hexagonal crystal structure wherein the early transition metal (M) atoms and the carbon/nitrogen (X) atoms make octahedral edge-sharing building blocks interleaved by pure A-group atomic layers [1]. MAX phases have good metallic properties high thermal shock resistance, electrical and thermal conductivity, damage tolerance, plastical deformation, whereas they are relatively soft and machinable [2–6]. Alike ceramics, they exhibit high melting temperature, high elastic rigidity, low density, creep and wear resistance, high elastic stiffness, high oxidation and corrosion resistance and can be used to form 2D-materials (MXenes) [7–22].
In recent years, MAX phase solid solutions attract a lot of interest due to the potential to tune the properties of their parent phases. In particular, the partial substitution of M-sites with different M-elements to form solid solutions is an effective way to modify the characteristics of MAX phase materials. Jiao et al. [23] studied theoretically the phase stability, mechanical properties and lattice thermal conductivity of the 413 MAX phase solid solutions (Nb1−x Ti x )4AlC3. This study reveals that mechanical properties of Nb4AlC3 are improved to some extent by the inclusion of Ti. An experimental study conducted by Gu et al. [24] shows that Ti strengthens Nb4AlC3. Usually, (Nb0.8Ti0.2)4AlC3 shows the highest flexural strength (508 ± 28 MPa) and fracture toughness (8.4 ± 0.2 MPa m1/2), growing by 47 and 18%, respectively. The Vickers hardness value of (4.4 ± 0.2) GPa for (Nb0.7Ti0.3)4AlC3 is 69% larger than that of Nb4AlC3. Exceptional wide-ranging properties constitute the (Nb1−x Ti x )4AlC3 solid solutions promising high-temperature structural materials. Lin et al. [25] investigated systematically the alloying effects on structural, magnetic, and electrical/thermal transport properties of Cr2−x M x GeC (M = Ti, V, Mn, Fe, and Mo). They showed that the alloying of M with the larger covalent radius than that of Cr increases lattice constants and unit cell volume of Cr2−x M x GeC, while the hexagonal ratio, electrical transport behavior, and the residual resistivity decreases with increasing x. Wang et al. [26] investigated the elastic stiffness and electronic properties of (\( {\text{M}}_{x} {\text{M}}_{2 - x}^{{\prime }} \))AlC (M and \( {\text{M}}^{\prime } \) are Ti, V, and Cr) solid solutions by ab initio calculations and found that an increment of the bulk modulus contributes to extra valence electrons occupying states involving M(\( {\text{M}}^{\prime } \)) d-Al p covalent bonding and metal–metal d–d bonding. Meng et al. [27] showed that 20% replacement of Ti with V in Ti2AlC enhances the Vickers hardness, flexural strength and shear strength by 29, 36 and 45% for (Ti0.8,V0.2)2AlC solid solution, respectively. Wan et al. [28] reported that Zr doping in Ti3SiC2 can improve the elastic modulus and strength at high temperatures, indicating a similar effect of Hf and Nb doping. Yeh et al. [29, 30] prepared (Ti1−x Nb x )2AlC, (Ti,V)2AlC and (Cr,V)2AlC solid solution via self-propagating high-temperature synthesis (SHS), and the strengthening effect was verified with the measured Vickers hardness and fracture toughness of the end members.
A new member in the MAX family, Zr3AlC2 was discovered in 2016 [31]. Among the 312 phases, Ti3AlC2 possesses excellent properties that make it a promising material for many industrial applications. Therefore, the solid solutions (Zr3−x Ti x )AlC2, mixing of Zr and Ti, should have properties better than that of many other MAX phases. Motivated by this, Zapata-Solvas et al. [32], very recently, successfully synthesized (Zr3−x Ti x )AlC2 MAX phases and calculated some physical properties with different x content. Here, we investigate the complementary structural and optical properties of (Zr3−x Ti x )AlC2 using density functional theory (DFT). In particular, the study of solid solutions (Zr3−x Ti x )AlC2 is carried out through the ground state electronic structure calculations by the use of the plane-wave pseudopotential method within DFT. The plan of the present paper is to examine the ground state properties including structural and optical properties of (Zr3−x Ti x )AlC2. The rest of the paper is arranged with three sections. In Sect. 2, a concise description of the computational method used in this study has been presented. The results on Fermi surface structural and optical properties of (Zr3−x Ti x )AlC2 are analyzed in Sect. 3. Finally, Sect. 4 consists of the main conclusions of this work.
2 Computational methods
The plane-wave pseudopotential method [33] based on DFT [34, 35] as implemented in the CASTEP code [36] is employed in the present study. Electronic exchange–correlation interaction is evaluated in accordance with the generalized gradient approximation (GGA) improved by Perdew–Burke–Ernzerhof (PBE) [37]. The Vanderbilt-type ultrasoft pseudopotentials [38] are used to assess the interactions between electrons and ion cores. The plane-wave energy cutoff is set at 550 eV and the k-points mesh [39] of 19 × 19 × 3 is selected for the Brillouin zone sampling. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm [40] is applied to optimize the total energy and internal forces. The convergence criteria for geometry optimization are: the difference in total energy per atom within 5 × 10−6 eV, the ionic Hellmann–Feynman forces within 0.01 eV/Å, maximum stress within 0.02 GPa, and the maximum ionic displacement within 5 × 10−4 Å. The effectiveness of atomistic simulations to explain defect processes has been discussed in early studies [41–48].
3 Physical properties
3.1 Structural properties
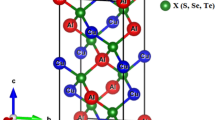
Consistently with their end members (i.e. Zr3AlC2 and Ti3AlC2) the MAX solid solutions (Zr3−x Ti x )AlC2 crystallize in the hexagonal structure in the space group P63/mmc (No. 194) [49, 50]. The transition metal atoms M(1) and M(2) occupy the Wyckoff positions 2a and 4f (zM ~ 0.12), respectively. The Al atoms reside in 2b atomic sites. The C atoms take up the 4f (zM ~ 0.07) atomic positions with different z-values. The unit cell of 312 MAX phases consists of two formula units (Fig. 1). The calculated lattice parameters and unit cell volumes along with experimental values are shown in Table 1. The present values agree quite well the measured values for two end products. The calculated lattice constants a and c of the new solid solutions (Zr3−x Ti x )AlC2 are plotted as a function of Ti content x in Fig. 2a. It is observed that both the lattice parameters a and c decrease radically as the Ti content x increases. Therefore, the volume of the unit cell must decrease with the increase of Ti content x and this is evident in Fig. 2b. The hexagonal ratio (c/a) showed in Fig. 2b increases rapidly as the Ti content x increases, which reveals that the values of c decrease less significantly than that of a. It means that the compressibility along c-axis decreases with increasing of Ti content x.

The unit cell of Zr3AlC2 as a structural model of 312 MAX phases

Structural properties of (Zr3−x Ti x )AlC2. a Lattice constants a and c; b hexagonal ratio c/a and unit cell volume V as a function of Ti content x. Experimental data adopted form Ref. [32]
3.2 Fermi surfaces
The Fermi surface topology has been calculated and presented in Fig. 3. The Fermi surfaces for all x contents are almost identical. Each Fermi surface consists of several tube-like sheets of hexagonal cross-sections centered along Γ–A direction. The first sheet is purely cylindrical shaped with hexagonal circumstance. The second sheet is much closer to the first sheet and has six appendices like fish tail along A–H directions. The third sheet is also cylindrical and it contains a sand-watch shaped tube along H–K directions at each corner of the Brillouin zone. The fourth sheet is prismatic-like. This sheet also contains an appendix shaped as a cone. An additional sheet appears in all compositions excepting two end members as two petals of a mike-flower at two sides of each corner of the Brillouin zone. All the sheets are separated from each other. The surfaces are formed mainly by the low-dispersive transition metal d-like bands, which are responsible for the conductivity in the compounds.

Fermi surfaces of (Zr3−x Ti x )AlC2 for x = 0.0, 0.5, 1.0, 1.5, 2.0, 2.5 and 3.0
3.3 Optical properties
In this section, we will discuss all the optical functions calculated for two end members of the MAX phase solid solutions (Zr3−x Ti x )AlC2 since such calculations are not allowed in the CASTEP code for disordered systems. The optical properties of solids may be described with the complex dielectric function, which is obtained as ε(ω) = ε 1(ω) + iε 2(ω); where ε 1(ω) is the real part of the complex dielectric function going behind the Kramers–Kronig relation and ε 2(ω) is the imaginary part. It is well established that the imaginary part ε2(ω) of the dielectric function leads to evaluate the remaining optical properties. The imaginary part of the dielectric function is evaluated from the momentum representation of matrix elements between the occupied and unoccupied wave functions with the selection rules [51] and can be expressed as:
where ω is the frequency of light, e is the charge of an electron, Ω is the unit cell volume, \( \varvec{u} \) is the unit vector defining the polarization of the incident electric field and \( \psi_{k}^{c} \) and \( \psi_{k}^{v} \) are the conduction and valence band wave functions at a given k, respectively. The imaginary part of dielectric function provides the real part trough Kramers–Kronig transformation. All other optical functions on the energy dependence of refractive index n(ω), extinction coefficient k(ω), absorption coefficient α(ω), energy-loss spectrum L(ω), optical conductivity σ(ω) and reflectivity R(ω) can be derived from the real and imaginary part of dielectric functions ε 1(ω) and ε2(ω) [52]. A Drude term [53] with unscreened plasma frequency of 3 eV and damping of 0.05 eV are included in all calculations because of the metallic nature of the compounds, which is confirmed in our band structure calculations. The smearing value of 0.5 eV is also used in each calculation of optical functions.
Dielectric function is a very common property of crystals. The calculated real part of the dielectric function for two end members of (Zr3−x Ti x )AlC2 is shown in Fig. 4a. It is seen that the real part ε 1(ω) of dielectric function in the range of ε 1 < 0 passes through zero from below. This is the indication of metallic characteristics of (Zr3−x Ti x )AlC2 MAX compounds. In the spectra for [100] polarization of both end compounds, the double peak structures are observed to be centered at 1.17 eV. At the same position, a single low peak arises for [001] polarization for x = 0 (Zr3AlC2), whereas a sharp dip is found for same polarization for x = 3 (Ti3AlC2). In the energy range of 0–5.4 eV, the spectra exhibit different features but at higher energies, the almost similar features are seen in the spectra for all polarizations. Indeed, the spectra disclose the dependence on polarization direction, which is the clear indication of anisotropy of optical properties of (Zr3−x Ti x )AlC2 MAX compounds in low energy range.

a Real and b imaginary part of dielectric functions of (Zr3−x Ti x )AlC2 for x = 0 and 3
The imaginary part ε2(ω) of the dielectric function is the fundamental issue of the optical functions for solids. The origin of the structures in ε2(ω) can shed light on the structures in the refractive index. For two end members of (Zr3−x Ti x )AlC2 the investigated imaginary part of the dielectric function is displayed in Fig. 4b. It is evident that the imaginary part ε2(ω) approaches zero from above, which insures the metallic characteristics of (Zr3−x Ti x )AlC2 as well. Only some numbers of low peaks are observed in the spectra of ε2(ω), which are due to the transition of orbital electrons. The peaks in the low energy region are caused by the intra-band transitions, but in the high energy region the peaks are due to the inter-band transition of electrons.
The proper knowledge on refractive index n(ω) of solids is important for perfect design of electronic appliances. Refractive index n(ω) is a dimensionless number that describe how electromagnetic radiation propagates through an optical medium. From the dispersion curves of n(ω) in Fig. 5a, the static values n(0) for two different polarization directions [100] and [001] are seen to be 84.52 and 84.85 for x = 0 (Zr3AlC2) and 84.51 and 85.92 for x = 3 (Ti3AlC2), respectively. A sharp rise is observed in all spectra within the moderate-infrared region, which is due to the intra-band transitions of electrons. The first peaks on the dispersion curves of n(ω) of Zr3AlC2 for incident light directions [100] and [001] arise at 0.68 and 0.44 eV, respectively. Such peak exists on n(ω) spectra of Ti3AlC2 for polarization [100] at around 0.64 eV, but no such peak arise for [001] polarization. A gradual decrease in spectra of n(ω) starts at 1.1 eV and reaches a minimum value of 0.02 at 9 eV. Then increases and attains a value of 0.85 at 20 eV. The refractive spectra show a slight anisotropy extended from 0.1 to 7.0 eV.

a Refractive index and b extinction coefficient of (Zr1−x Ti x )3AlC2 for x = 0 and 3
The extension coefficient k(ω) is more significant phenomena in metals, which estimates the amount of absorption loss when the electromagnetic wave propagates through the optical medium. For two different polarization directions the extinction coefficient of two end members of (Zr3−x Ti x )AlC2 is presented in Fig. 5b. The calculated extinction coefficient shows small anisotropy up to 13.6 eV. In both end compounds, the spectra for two polarization directions exhibit sharp rise below 2 eV, which is caused by the intra-band transitions of electrons. The local maxima on the extinction coefficient curves correspond to the zero value of the real part of dielectric function.
The absorption coefficient estimates how far light of a particular wavelength (energy) can penetrate into the material before being absorbed and affords information related to the optimum solar energy conservation efficiency. The absorption coefficient of Zr3AlC2 and Ti3AlC2, end members of (Zr3−x Ti x )AlC2 MAX compounds are plotted in Fig. 6a. The absorption spectra commence at zero photon energy due to the metallic nature of the compounds. For both the polarization directions, the absorption spectra increases with the increase of photon energy to achieve the highest values within the energy range 4.5–5.5 eV for Zr3AlC2 and within 5–6 eV for Ti3AlC2. Further increase of photon energy decreases the absorption coefficient. The calculated absorption spectra show a small amount of anisotropy up to the photon energy of 13 eV.

a Absorption coefficient and b energy loss function of (Zr1−x Ti x )3AlC2 for x = 0 and 3
The energy loss function L(ω) describes the energy loss of fast electron traveling inside the material. The energy loss spectra shown in Fig. 6b exhibit no distinct peak in the energy range of 0–10 eV due to large value of the imaginary part of the dielectric function [54]. A large peak on each spectrum is observed within the energy range of 10–14 eV, which is associated with the plasma oscillation. The highest peak of energy loss spectrum refers a characteristic frequency, known as bulk plasma frequency, at which the real part of dielectric function vanishes and the material changes from metallic to dielectric response [53, 55]. From the energy loss spectra, the bulk plasma frequency of Zr3AlC2 and Ti3AlC2 for the polarization directions [100] and [001] are found to be 11.9, 13.4 and 10.8, 13.5 eV, respectively, which correspond to the rapid decline of reflectivity given in Fig. 7a.

a Reflectivity and b optical conductivity of (Zr3−x Ti x )AlC2 for x = 0 and 3
Reflectivity is the ratio of the energy associated with the wave reflected from a surface to the energy possessed by the wave striking the surface. The optical reflectivity R(ω) calculated here is plotted in Fig. 7a. The spectra of reflectivity exhibit a radical increase in the moderate infrared region as well as in the ultra-violet region to attain the highest values of 92.7–96.7%. The reflectivity spectra show no significant change in the visible light region (~1.8–3.1 eV) and the amount of reflectivity exceeds 47%, which should make two end members of (Zr3−x Ti x )AlC2 candidate materials for coating to reduce solar heating [56]. Additionally, the two end MAX compounds Zr3AlC2 and Ti3AlC2 should appear as metallic gray due to their nearly constant reflectivity in the visible light region. The investigated reflectivity spectra show a reasonable amount of anisotropy within the photon energy range of 0.5–14 eV.
Optical conductivity signifies the electrical conductivity in the presence of an alternating electric field; here the term ‘optical’ covers the whole frequency range, and is not limited only to the visible region of the light. The real part of optical conductivity represents the in-phase current, which causes the resistive joule heating, whereas the imaginary part involves in the π/2 out-of-phase inductive current. Here we have calculated the real part of optical conductivity and shown in Fig. 7b. The highest peak on the spectra of optical conductivity indicates that Zr3AlC2 is expected to be highly electrically conductive when incident photon along [100] direction has energy equal to 3.1 eV and when along [001] direction has 4 eV. Similarly, Ti3AlC2 will be highly electrically conductive when incident photon has 3 and 3.3 eV for [100] and [001] direction, respectively. The calculated optical conductivity exhibit a sensible amount of anisotropy within the photon energy range of 0.2–7 eV. The peak’s height of optical conductivity for incident photon direction [001] is seen to be larger than that for [100] direction in case of both end members of (Zr3−x Ti x )AlC2.
4 Conclusions
In the present study, DFT calculations are employed to predict the lattice parameters, and optical functions of the recently synthesized (Zr3−x Ti x )AlC2 MAX phases. The lattice parameters are in excellent agreement with the recently determined results [32]. The Fermi surfaces consist of low-dispersive transition metal d-like bands, which play a vital role for the conductivity in the compounds. The optical properties are deemed to be polarization dependent. The bulk plasma frequency of Zr3AlC2 and Ti3AlC2 for the light directions [100] and [001] are estimated from the energy loss spectra and found to be 11.9, 13.4 and 10.8, 13.5 eV, respectively, which correspond to the rapid fall of reflectivity. The reflectivity spectra in the visible light region hint that Zr3AlC2 and Ti3AlC2 are candidate materials for coatings to reduce solar heating.
References
M.W. Barsoum, Prog. Solid State Chem. 28, 201 (2001)
H. Yoo, M.W. Barsoum, T. El-Raghy, Nature 407, 581 (2000)
M.W. Barsoum, T. El-Raghy, J. Am. Ceram. Soc. 79, 1953 (1996)
T. El-Raghy et al., J. Am. Ceram. Soc. 82, 2855 (1999)
M.W. Barsoum, L. Farber, T. El-Raghy, Metall. Mater. Trans. A 30, 1727 (1999)
Z.M. Sun et al., Mater. Trans. 47, 170 (2006)
P. Finkel, M.W. Barsoum, T. El-Raghy, J. Appl. Phys. 87, 1701 (2000)
M.W. Barsoum, Physical properties of the MAX phases, Encyclopedia of Materials: Science and Technology (Elsevier, Amsterdam, 2009)
M. Radovic et al., J. Alloys Compds. 361, 299 (2003)
C.J. Gilbert et al., Scri. Mater. 238, 761 (2000)
M. Sundberg et al., Ceram. Int. 30, 1899 (2004)
V.D. Jovic et al., Corr. Sci. 48, 4274 (2006)
A. Talapatra, T. Duong, W. Son, H. Gao, M. Radovic, R. Arroyave, Phys. Rev. B 94, 104106 (2016)
J. Zhu, A. Chroneos, J. Eppinger, U. Schwingenschlögl, Appl. Mater. Today 5, 19 (2016)
M. Dahlqvist, J. Phys. Condens. Matter 28, 135501 (2016)
D. Horlait, S. Grasso, A. Chroneos, W.E. Lee, Mater. Res. Lett. 4, 137 (2016)
M. Khazaei, A. Ranjbar, M. Ghorbani-Asl, M. Arai, T. Sasaki, Y.Y. Liang, S. Yunoki, Phys. Rev. B 93, 205125 (2016)
A. Chroneos, S.C. Middleburgh, A. Chroneos, W.E. Lee, Sci. Rep. 6, 18829 (2016)
A.S. Ingason, M. Dahlqvist, J. Phys. Condens. Matter 28, 433003 (2016)
D. Cakir, C. Sevik, O. Gulseren, F.M. Peeters, J. Mater. Chem. A. 4, 6029 (2016)
J. Zhu, A. Chroneos, U. Schwingenschlögl, Phys. Status Solidi RRL 9, 726 (2016)
B. Anasori, M. Dahlqvist, J. Halim, E.J. Moon, J. Lu, B.C. Hosler, E.N. Caspi, S.J. May, L. Hultman, P. Eklund, J. Rosen, M.W. Barsoum, J. Appl. Phys. 118, 094304 (2015)
Z.Y. Jiao, T.X. Wang, S.H. Ma, J. Alloys Compd. 687, 47 (2016)
J. Gua, L. Pan, J. Yang, L. Yu, H. Zhang, W. Zou, C. Xu, C. Hu, T. Qiu, J. Euro. Ceram. Soc. 36, 1001 (2016)
S. Lin, Y. Huang, L. Zu, X. Kan, J. Lin, W. Song, P. Tong, X. Zhu, Y. Sun, J. Alloys Comp. 680, 452 (2016)
J. Wang, Y. Zhou, J. Phys. Condens. Matter 16, 2819 (2004)
F. Meng, Y. Zhou, J. Wang, Scri. Mater. 53, 1369 (2005)
D.T. Wan et al., J. Am. Ceram. Soc. 93, 1749 (2010)
C.L. Yeh, J.H. Chen, Ceram. Int. 37, 3089 (2011)
C.L. Yeh, W.J. Yang, Ceram. Int. 39, 7537 (2013)
T. Lapauw, J. Halimc, J. Lu, T. Cabioc’h, L. Hultman, M.W. Barsoum, K. Lambrinou, J. Vleugels, J. Euro. Ceram. Soc. 36, 943 (2016)
E. Zapata-Solvas, M.A. Hadi, D. Horlait, D.C. Parfitt, A. Thibaud, A. Chroneos, W.E. Lee, J. Appl. Phys. submitted
R. Martin, Electronic Structure: Basic Theory and Practical Methods (Cambridge University Press, Cambridge, 2004)
P. Hohenberg, W. Kohn, Phys. Rev. 136, B864 (1964)
W. Kohn, L.J. Sham, Phys. Rev. 140, A1133 (1965)
S.J. Clark, M.D. Segall, C.J. Pickard, P.J. Hasnip, M.I.J. Probert, K. Refson, M.C. Payne, Z. Krist. 220, 567 (2005)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
D. Vanderbilt, Phys. Rev. B 41, 7892 (1990)
H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13, 5188 (1976)
T.H. Fischer, J. Almlof, J. Phys. Chem. 96, 9768 (1992)
R.W. Grimes, G. Busker, M.A. McCoy, A. Chroneos, J.A. Kilner, S.P. Chen, Berich. der Bunsen. für phys. Chem. 101, 1204 (1997)
A. Chroneos, C. Jiang, R.W. Grimes, U. Schwingenschlögl, H. Bracht, Appl. Phys. Lett. 95, 112101 (2009)
D. Rupasov, A. Chroneos, D. Parfitt, J.A. Kilner, R.W. Grimes, S.Y. Istomin, E.V. Antipov, Phys. Rev. B 79, 172102 (2009)
S.T. Murphy, A. Chroneos, C. Jiang, U. Schwingenschlögl, R.W. Grimes, Phys. Rev. B 82, 073201 (2010)
A. Chroneos, C.A. Londos, E.N. Sgourou, J. Appl. Phys. 110, 093507 (2011)
I.D. Seymour, A. Chroneos, J.A. Kilner, R.W. Grimes, Phys. Chem. Chem. Phys. 13, 15305 (2011)
I.D. Seymour, A. Tarancon, A. Chroneos, D. Parfitt, J.A. Kilner, R.W. Grimes, Solid State Ion. 216, 41 (2012)
M.J.D. Rushton, A. Chroneos, Sci. Rep. 4, 6068 (2014)
T. Lapauw, J. Halim, J. Lud, T. Cabioc’h, L. Hultman, M.W. Barsoum, J. Vleuge, K. Lambrinou, J. Eur. Ceram. Soc. 36, 1847 (2016)
M. Pietzka, J. Schuster, J. Phase Equilib. 15, 392 (1994)
D.C. Harris, M.D. Bertolucci, Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy, 1st edn. (Dover Publications, USA, 1989)
M.A. Hadi, R.V. Vovk, A. Chroneos, J. Mater. Sci: Mater. Electron. 27, 11925 (2016)
R. Saniz, L.H. Ye, T. Shishidou, A. Freeman, J. Phys. Rev. B 74, 014209 (2006)
J.P. Watt, L. Peselnick, J. Appl. Phys. 51, 1525 (1980)
J.S. de Almeida, R. Ahuja, Phys. Rev. B 73, 165102 (2006)
S. Li, R. Ahuja, M.W. Barsoum, P. Jena, B. Johansson, Appl. Phys. Lett. 92, 221907 (2008)
Acknowledgements
A. C. is grateful for funding from the Lloyd’s Register Foundation, a charitable foundation helping to protect life and property by supporting engineering-related education, public engagement and the application of research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hadi, M.A., Panayiotatos, Y. & Chroneos, A. Structural and optical properties of the recently synthesized (Zr3−x Ti x )AlC2 MAX phases. J Mater Sci: Mater Electron 28, 3386–3393 (2017). https://doi.org/10.1007/s10854-016-5933-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-016-5933-z