
Abstract
In this work, new bio-based copoly(ester amide)s were synthesized by a two-step melt polycondensation process, using 2,5-furanedicarboxylic acid dimethyl ester (DMFDC), 1,3-propanediol (PDO), and 1,3-diaminopropane (DAP), with different DAP content. The chemical structure of the obtained poly(trimethylene 2,5-furandicarboxylate)-co-poly(propylene furanamide) (PTF-co-PPAF) copolymers was confirmed by nuclear magnetic resonance (1H NMR) and Fourier-transform infrared (FTIR) spectroscopy. Gas chromatography/mass spectrometry was used to provide more details of the polycondensation process. Thermal properties of the obtained materials were characterized by means of differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and dynamic–mechanical thermal analysis (DMTA). The copolymers were amorphous and their glass transition temperature increased with the increase in the poly(propylene furanamide) (PPAF) content. The synthesized PTF-co-PPAF copolymers exhibited improved thermal and thermo-oxidative stability up to 300 °C. In addition, from the performed mechanical tests, it was found that along with the increase in PPAF content, Young's modulus increased, while at the same time, the value of elongation at break decreased.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Polyamides based on renewable raw materials have aroused the interest of scientists for many years. Already in the 40 s of the 20th century, polyamide 11 was first synthesized from castor oil (Rilsan) [1]. 20 years later, the first syntheses of polyamides based on 2,5-furanedicarboxylic acid (FDCA) took place [2]. Currently, there are a number of polyamides obtained from sebacic acid on the market (Vestamid from Evonik, EcoPaXX from DSM) [3,4,5].
The decreasing resources of fossil fuels and the growing ecological awareness of the society have only increased the interest in the use of renewable raw materials in the production of polymers in recent years. Among them, FDCA attracts special attention. Due to its structure, similar to terephthalic acid (TPA), in 2004, it was recognized as one of the 12 most promising compounds of plant origin for the synthesis of polymeric materials [6]. In later years, numerous studies showed that it is actually a monomer highly suitable for the synthesis of polyesters [7,8,9,10,11,12], polyurethanes [13], or various types of copolymers [14,15,16,17]. As Terzopoulou and others [18] pointed out, there is a growing number of works on furan-based copolyesters, which is related to their easy synthesis and interesting, various properties. Moreover, the use of FDCA allows obtaining materials with better properties than their counterparts based on TPA. An example is poly(ethylene 2,5-furanoate) (PEF), which is characterized by much better barrier properties (11 × lower O2 permeability, 19 × lower CO2 permeability) than widely used, especially in the packaging industry, polyethylene terephthalate (PET) [19, 20].
In the case of polyamides, difficulties have been encountered during the syntheses with FDCA, so there is still relatively little work describing the properties of these bio-based materials. The low molecular weights of the end products, resulting from the decarboxylation of FDCA, make it difficult to obtain materials with promising properties [21]. The work, therefore, focused mainly on the development of the synthesis method itself. Thus, one can find publications describing, among others, solution polymerization of the diamines with various FDCA derivatives [22,23,24,25,26], direct polycondensation method [25, 27, 28], interfacial polymerization [25, 26, 29, 30] as well as solid-state polymerization [31, 32]. The by-products produced during the syntheses make that a method that would allow obtaining a large amount of pure material with high molecular weight is still sought after. In recent years, Jiang et al. [33, 34], in order to obtain FDCA-based polyamides, used the enzyme Novozym 435. The enzymatic polymerization, due to the milder reaction conditions (lower temperature and pressure), appeared to be a promising method, but even in its case, the reaction efficiency did not exceed 50% [34]. Some other researchers also described enzymatic polymerization using the same enzyme, to obtain furan-based polyamides [35]. To overcome the low molecular weight problems, it seemed promising to synthesize copolyamides and copoly(ester amide)s (PEAs) based on FDCA [36,37,38,39]. The resulting intermolecular hydrogen bonds, however, resulted in the deterioration of the thermal and mechanical properties of the materials [38]. To solve this problem, Kluge et al. [40] used a diol containing two internal amide bonds. However, due to long aliphatic chains, the obtained materials had relatively low Young's modulus values.
On the basis of the available literature, it can be seen that in numerous studies, there were difficulties in obtaining high molecular furan-based polyamides, copolyamides, and copoly(ester amide)s. Despite many challenges encountered in the synthesis of these materials, interest in them continues to grow. Unfortunately, most papers lack detailed studies of polyamides and copoly(ester amide)s, especially regarding their mechanical properties. Usually, too little of the end product is obtained to test its tensile strength, especially on dumbbell-shaped specimens shaped by injection molding. Hence, in this study, four copoly(ester amide)s (PEAs) based on 2,5-furanedicarboxylic acid dimethyl ester (DMFDC), 1,3-propanediol (PDO), and 1,3-diaminopropane (DAP) with different DAP content were synthesized and characterized. During each synthesis, about 200 g of the final product was obtained. The chemical structure, thermal and mechanical properties of the obtained PEAs were investigated and compared to the synthesized poly(trimethylene 2,5-furan dicarboxylate) (PTF).
Materials and methods
Preparation procedure of copoly(amide-esters)
Neat poly(trimethylene 2,5-furanoate) (PTF) and copoly(ester amide)s based on dimethyl furan 2,5-dicarboxylate (DMFDC) were prepared by a two-stage melt polycondensation method (Fig. 1). In the first step, the transesterification reaction between DMFDC (DMFDC, 99%, Henan Coreychem Co., Ltd., Zhengzhou, China), 1,3-propylene glycol (bio-PDO, DuPont Tate & Lyle BioProducts, Loudon, USA), and 1,3-diaminopropane (DAP, > 99%, Sigma-Aldrich), in the presence of titanium (IV) butoxide (Fluka® Analytical) as a catalyst, was carried out. The molar ratio of diester to glycol was 1:2. During this step, methanol was collected as a by-product. The transesterification reaction was conducted at 160–190 °C for 2 h. When almost all theoretical amount of methanol was distilled off (~ 90%), the temperature was increased up to 220 °C and Irganox 1010 (Ciba-Geigy, Switzerland) as a thermal stabilizer along with the second portion of catalyst was introduced to the reactor. Then, the pressure of the reaction was gradually reduced below 20 Pa and the temperature was increased up to 240 °C. The second stage, melt polycondensation, was finished when melt reaches a specified value of viscosity, which was monitored by an increase in the stirrer torque. The prepared materials were extruded from the reactor as a strand into the water bath, granulated, and dried. The copolymers were transparent with an orange tint and became darker in color as the amount of PPAF content increased. A total of 5 materials were synthesized: 4 copoly(ester amide)s with various molar ratios of PTF to PPAF and neat PTF.

Synthesis of PTF-co-PPAF copolymers
Methods
The chemical structures of poly(trimethylene 2,5-furandicarboxylate)-co-poly(propylene furanamide) (PTF-co-PPAF) copolymers were studied by 1H NMR. Prior to the experiment, all samples were subjected to continuous Soxhlet extraction with methanol for 24 h, to remove unreacted monomer and possible low molecular weight degradation products. 1H NMR spectra were recorded on a Bruker spectrometer operated at 400 MHz, using chloroform-d (CDCl3) with a few drops of trifluoroacetic acid (CF3COOH) as solvents. Tetramethylsilane (TMS) was used as an internal chemical shift reference. To calculate the real fractions of PPAF segments in the synthesized copolymers, the determined integral intensities of the characteristic peaks from 1H NMR data were used.
The infrared spectra were acquired at room temperature with a Nicolet 380 ATR-FTIR spectrometer (Thermo Scientific, USA). 16 scans were averaged for each sample in the range of 4000–400 cm−1. Measurements were made on dumbbell-shaped samples obtained by injection molding. The injection parameters are described in the description of the mechanical properties test method.
The intrinsic viscosity (IV) of the series of copoly(ester amide)s was determined in the mixture of phenol/1,1,2,2-tetrachloroethane (60/40 by weight) at 30 °C. The concentration of the polymer solution was 0.5 g/dL. The measurement was performed using a capillary Ubbelohde viscometer (type Ic, K = 0.03294).
The density of samples was determined on the hydrostatic scales (Radwag WPE 600C, Radom, Poland) at room temperature (23 °C), calibrated according to the standards with a known density. For each material, 10 measurements were performed.
Control over the polycondensation process was carried out by gas chromatography coupled with mass spectrometry. Qualitative and quantitative analysis was performed with the GC–MS ThermoQuest apparatus, equipped with a Voyager detector and a DB-5 column (filled with phenylmethylsiloxanes, 30 m × 0.25 mm × 0.5 um). The analysis parameters were as follows: helium flow 1 ml/min, inlet temperature 150 °C, furnace temperature isothermally for 2.5 min at 50 °C, then an increase at a rate of 10 °C/min to 300 °C. Distillate compositions were calculated using the internal normalization method, taking into account response factors.
Thermal properties of the copolymers were determined by differential scanning calorimetry (DSC), dynamic mechanical thermal analysis (DMTA), and thermogravimetric analysis (TGA). Differential scanning calorimetry (DSC) measurements of PTF-co-PPAF copolymers were conducted on a DSC 204 F1 Phoenix (Netzsch, Selb, Germany). About 10 mg of a sample was encapsulated in an aluminum crucible (pan) and heated from − 75 to 250 °C at a scan rate of 10 °C/min. Measurements were performed under a nitrogen-rich atmosphere using a method with a heat/cool/heat cycle. Dynamic mechanical thermal analysis (DMTA) was performed using a TA Instruments DMA Q800. All samples were tested at a frequency of 1 Hz and a heating rate of 3 °C/min. The storage modulus (E'), loss modulus (E''), and tan δ were determined as a function of temperature. The thermal and thermo-oxidative stability of the copoly(ester amide)s was examined via Thermogravimetric Analysis (TGA 92–16.18 Setaram). PTF-co-PPAF samples were heated in an inert atmosphere (nitrogen) and oxidizing atmosphere (N2: O2 = 80: 20 vol. %) at a flow rate of 20 mL/min. The temperature range of 20–700 °C and the heating rate of 10 °C/min were set for this study.
Dumbbell-shaped samples (type A3) for mechanical tests were obtained by injection molding using a BOY 15 injection molding machine (Dr BOY GmbH and Co., Germany). The following injection parameters were used: injection pressure 85 MPa, melt temperature 195 °C, mold temperature 30 °C, holding down pressure of 30 MPa for 6 s, and cooling time of 15 s. The mechanical properties were appraised via a universal tensile test machine (Autograph AG–X plus, Shimadzu), equipped with an optical extensometer according to EN ISO 527. The strain rate was set to 1 mm/min up to 1% of strain in order to calculate the tensile modulus, and then, the strain rate was set to 5 mm/min until the sample was broken. The average of six measurements was calculated for each sample.
The hardness of investigated materials was measured on Shore D tester (Karl Frank GmbH, Type 104, Germany) according to a standard DIN 53,505 and ISO 868.
Results
Structure and composition
PTF-co-PPAF copoly(ester amide)s were synthesized by a two‐stage melt polycondensation method. This method is particularly attractive, as it enables to obtain a substantial amount of material. In Table 1, the characteristic properties of the obtained copolymers are presented. Depending on the copolymer composition, the materials are characterized by the intrinsic viscosity number (IV) from 0.651 to 0.799 dl/g for PTF-co-PPAF 1/0.16. The IV closely depends on the molecular weight; hence, it can be concluded that the highest molecular weights were obtained for the middle compositions. The values of IV for all obtained materials were relatively high and were comparable to those for the bottle grade PET that has an IV of 0.7–0.78 dl/g [41]. However, neat PTF homopolymer had a lower IV value than in our previous studies (0.800 dl/g) [42]. The IV value may be influenced by the method of synthesis, reaction conditions, and the type of catalyst used [43]. In our case, the synthesis conditions were identical; hence, the differences in the IV values may result from differences in molecular weights. The densities of the obtained copolymers were slightly lower than that of neat polyester and ranged from 1.28 to 1.33 g/cm3.
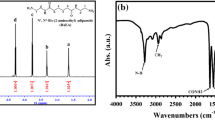
The chemical structure and composition of the obtained copoly(ester amide)s were confirmed by 1H NMR and FTIR spectroscopies. The 1H NMR spectra of the PTF-co-PPAF copolymers are shown in Fig. 2. All signals corresponding to the PTF unit are clearly visible. The intense peak at 7.24 ppm (a) is associated with the protons of the furan ring. This signal overlaps slightly with the resonance from the solvent (CDCl3). In addition, two more intense peaks come from the PTF unit: a triplet located at 4.52 ppm (b1) and a triplet located at 2.27 ppm (c), which corresponds to the 6 protons of the glycol subunit. For PEAs, some new signals associated with ester − amide sequences appeared. New peaks occurring at 3.63 ppm and 3.97 ppm can be attributed to the methyl group protons linked to the nitrogen atom and amide protons, respectively. It is worth noting that in the case of PEAs, the amide protons may be delocalized. As noted by Wilsens et al. [38], between the oxygen heteroatom in the furan ring and the hydrogens of the amide bonds, intermolecular hydrogen bonds may form. However, this would result in the loss of resonance corresponding to the amide protons. On the basis of the obtained 1H NMR results, it can be concluded that FDCA-based copoly(ester amide)s have been successfully synthesized.

Structure and H NMR spectra of PTF-co-PPAF copoly(ester amide)s
To estimate the actual mole fractions of the PPAF units, the copolymers compositions (WPPAF) were calculated according to Eq. (1):
where Ie and Ic are the internal signal intensities for the corresponding peak at 3.63 ppm and 2.26 ppm, respectively.
Calculated molar fractions are summarized in Table 1. The mole fractions of PPAF units estimated from 1H NMR are similar to the theoretical values. For the PTF-co-PPAF 1:0.06 and PTF-co-PPAF 1/0.5 copolymers, the calculated mole fractions of PPAF are slightly lower than the theoretical values, which may be due to the by-products formed and the inferior molecular weights of these materials. The lower molecular weights of these copolymers are also indicated by the minor IV values, compared to the other materials obtained. Moreover, the lower molecular weight of the PTF-co-PPAF 1/0.06 copolymer is also confirmed by the number of weak signals below 2 ppm, arising from the end groups (Fig. 2).
The FTIR spectra also confirm the successful synthesis of PTF-co-PPAF copoly(ester amide)s. Figure 3 shows the spectra of 1,3-diaminopropane, neat PTF, and PTF-co-PPAF copolymers. The diagram shows characteristic bands corresponding to the amine groups at the wavenumbers of 3380, 1665, and 1600 cm−1. The 3380 cm−1 band is responsible for the stretching vibrations of the N–H groups, while the 1665 and 1600 cm−1 bands are responsible for the bending vibrations of the N–H groups [44]. As the content of 1,3-diaminopropane increases, the intensity of the bands corresponding to the amine groups increases. Moreover, the signals corresponding to the furan ring are also visible. The strong absorption peaks at 1575–1580 cm−1 and 3125 cm−1 are attributed to the C=C stretching bonds and C–H stretching bonds, respectively. Furthermore, absorption assigned to=C–O–C=antisymmetric stretching and=C–O–C=ring vibrations appear at 1261–1266 cm−1 and 1211–1219 cm−1, respectively. The strong absorption peaks at 1710 cm−1 are attributed to C=O stretching vibration in PTF and PPAF segments. The signals around 2965 cm−1 corresponding to the CH2 groups of PDO and PPAF moieties are visible. The furan ring bending peaks are featured in three locations, 962, 822, 760 cm−1, while the breathing peaks are observed at around 1017 cm−1 [11, 45]. The obtained results confirm the incorporation of the amine into the copolymer structure.

FTIR spectra of 1,3-diaminopropane, neat PTF, and PTF-PPAF copolymers
The compositions of distillates from the reaction progress tests are presented in Table 2. Based on the chromatographic analyses of the distillates obtained during the syntheses of copoly(ester amide)s based on PDO, DAP, and DMFDC, it was found that DAP reacts completely and is not present in the distillates. In the first stage of polycondensation, the main component of the distillate was methanol and 1-butanol. In the second stage of polycondensation, the main component of the distillate was 1,3-propanediol; moreover, 1-butanol, ethylene glycol, 3-methoxy-1-propanol, and methanol were formed to a small extent. The analyses performed during the syntheses confirmed that the reaction was proceeding as assumed. The resulting distillates could be reused, also for subsequent syntheses.
Thermal properties
The synthesized copoly(ester amide)s were characterized by DSC, DMTA, and TGA to study the influence of the PPAF content on their glass transitions, thermal effects, as well as their thermal and thermo-oxidative stability. The results of DSC measurements, as well as the graph of the dependence of the PPAF content on the glass transition temperature, are shown in Fig. 4. As noted in the literature, polyamides and copoly(ester amide)s based on furandicarboxylic acid are usually amorphous [30, 36, 37, 46]. Moreover, furan-based polyesters with shorter aliphatic chains (up to 5 methylene groups), including PTF, are characterized by slow crystallization [47, 48]. The greater stiffness of the furan ring compared to benzene, caused by the smaller bond angles, makes the ring movement more difficult. In addition, the polarization and the nonlinear axis of rotation further frustrate the furan ring-flipping [7, 18, 49,50,51]. Also in our case, PTF-co-PPAF copolymers were amorphous, as no crystallization or melting peaks were observed on DSC thermograms. Along with the increase in PPAF mole fraction, a clear increase in the glass transition temperature was noticed, due to the rigidifying effect of intermolecular hydrogen bonds of amide groups. The obtained materials were characterized by Tg ranging from 55.9 to 73.9 °C.

Differential scanning calorimetry (DSC) thermograms for PTF and PTF-co-PPAF copoly(ester amide)s recorded during 2nd heating (a) and cooling (b), and graph of the dependence of the PPAF units content on the Tg (c)
The DMTA results are in the agreement with DSC data. The changes of storage modulus (E’), loss modulus (E”), and loss tangent (tan δ) as a function of temperature for the neat PTF and PTF-co-PPAF copolymers are presented in Fig. 5. The first drop in E' is related to the α-relaxation process. The glass transition temperatures increased along with the increasing PPAF mole fraction, which can be seen from the peak position in the loss module (E") and the tan δ traces. In order to better present the dependence of the PPAF units content on Tg, the glass transition temperatures determined by the DMTA method are summarized in the Table 3. The Tg of the obtained materials, determined from the curves of the storage modulus, has the lowest values (61–73 °C), while the highest values were recorded from the tanδ curves (68–81 °C). As can be seen in Fig. 5a, in the case of PTF, an additional peak appeared at 147 °C, which may be related to cold crystallization during the heating process. During DMTA measurements, the samples were heated slower than in the DSC method; hence, no cold crystallization was observed in Fig. 4a.

The storage and loss modulus (a) and normalized loss tangent (b) as a function of temperature for PTF-co-PPAF copoly(ester amide)s
Thermal stability of the synthesized copoly(ester amide)s was determined by thermogravimetric analysis (TGA) in an oxidizing and inert atmosphere, and the obtained results are depicted in Fig. 6. The temperatures corresponding to 5, 10, and 50% mass loss, as well as the temperatures corresponding to the maximum of mass losses, are presented in Table 4. Generally, the FDCA monomer is less stable than the terephthalic one [52]. Despite this, all obtained copolymers are stable up to approximately 300 °C in both oxidized and inert atmospheres, which is a satisfactory result. In the case of measurements carried out in an oxidizing environment, there is a two-stage degradation process. The first step principally associated with the polymer backbone occurs in the temperature range of 300–400 °C. As the content of PPAF increases, an increase in decomposition onset temperature was observed. Neat PTF showed a 5% mass loss at a temperature of 10 °C lower than the PTF-co-PPAF 1/0.25 copolymer. In the case of the second step attributed to the decomposition of the residue, the temperatures corresponding to the maximum of mass losses are visibly higher for the copolymers with higher PPAF content. In an inert atmosphere, the degradation process of the obtained materials proceeds in one step. Among synthesized copolymers, PTF-co-PPAF 1/0.5 was found to be the least thermally stable. As it is known, thermal stability is greatly influenced by the molecular weight and the number of end groups [30, 53]. It is, therefore, possible that in the case of the PTF-co-PPAF 1/0.5 copolymer, a greater number of low molecular weight products were obtained, which had a negative effect on the thermal stability of this material. This is also confirmed by the results obtained from 1H NMR, where the calculated mole fraction of PPAF in the case of this copoly(ester amide) was almost 5% lower than the assumed value.

TGA thermograms of PTF-co-PPAF copolymers polymers under thermo-oxidative (a) and inert (b) atmosphere
Tensile properties
The mechanical properties of the obtained copoly(ester amide)s were studied and further discussed. The representative stress–strain curves are shown in Fig. 7. In addition, the Young's modulus (E), stress at break (σb), elongation at break (εb), and Shore D hardness of copolymers were determined, and their results are summarized in Table 5. The content of PPAF units did not affect the hardness of copolymers but had a significant impact on the stiffness and tensile strength of the materials. All synthesized materials were characterized by hardness of 72 ShD. However, as expected, significant changes in the values of Young's modulus were noticed. For example, Young's modulus for PTF-co-PPAF 1/0.50 was more than 40% higher than the value obtained for neat polyester. The reasons are microstructure and strong intermolecular hydrogen bonding of the amide groups [54]. The tensile strength of the copolymers is lower than that of PTF. A similar relationship can be found in the literature for poly(ester amide)s elastomers [55]. All of the obtained materials showed a low value of elongation at break (up to 3,13%), which decreased along with increasing PPAF content. It is worth mentioning that the high stiffness of the obtained copolymers and their low elongation at break is largely influenced by the presence of the furan ring. As is known, furan-based polyesters are characterized by greater stiffness and tensile strength than their terephthalic counterparts [49, 56]. This is due to increased chain rigidity, associated with lower bond angles of 2,5-furandicarboxylic acid with respect to terephthalic acid.

The representative stress–strain curves of neat PTF and PTF-co-PPAF copolymers
Conclusions
Copoly(ester amide)s are interesting materials due to the combination of the main characteristics of polyamides, such as stiffness, excellent thermal and mechanical properties, with the biocompatibility of polyesters. In this work, four poly(ester amide)s with different PPAF content were successfully synthesized employing a two-step melt polycondensation method. The products were verified by 1H NMR, GC–MS, and FTIR spectroscopies, and it can be concluded that the amide moieties were auspiciously incorporated into the PTF backbone. The obtained materials were completely amorphous, due to the stiffening effect of the furan ring. The content of PPAF had a significant influence on the glass transition temperature of the copolymers. As the PPAF mole fraction increased, the Tg shifted toward higher values. All copolymers were characterized by good thermal and thermo‐oxidative stability, and the increase in the PPAF content had a positive effect on the values of decomposition temperatures of the copoly(ester amide)s. Incorporation of 1,3-diaminopropane significantly affected the mechanical performance, providing high Young’s modulus but limiting elongation at break. From the aforementioned results, we conclude that the furan-based copoly(ester amide)s can be found as promising materials for bio-based copolymers production with good strength properties and satisfactory thermal stability. Good properties of copoly(ester amide)s result, among others, from the strong intermolecular hydrogen bondings of the amide groups (–NHCO–). In future, it could be considered to obtain and characterize in terms of thermal and mechanical properties, of semi-crystalline furan-based copoly(ester amide)s.
References
Genas M (1962) Rilsan (Polyamid 11), synthese und eigenschaften. Angew Chemie 74:535–540. https://doi.org/10.1002/ange.19620741504
Hopff VH, Krieger A (1961) Über polyamide aus heterocyclischen dicarbonsäuren. Die Makromol Chem 47:93–113. https://doi.org/10.1002/macp.1961.020470109
Winnacker M, Rieger B (2016) Biobased polyamides: recent advances in basic and applied research. Macromol Rapid Commun 37:1391–1413. https://doi.org/10.1002/marc.201600181
VESTAMID® TERRA TECHNICAL PROPERTIES. https://www.vestamid.com/en/products-services/VESTAMID-terra/technical-properties. Accessed 4 Mar 2021
EcoPaXX®: The green performer The bio-based carbon neutral polyamide. https://www.dsm.com/content/dam/dsm/ecopaxx/en_US/documents/DSMEP_BrochureEcoPaXX_finalLR.pdf. Accessed 4 Mar 2021
Werpy T, Petersen G (2004) Top Value Added Chemicals from Biomass: Volume I -- Results of Screening for Potential Candidates from Sugars and Synthesis Gas. Golden, CO (United States)
Zubkiewicz A, Paszkiewicz S, Szymczyk A (2021) The effect of annealing on tensile properties of injection moThe effect of annealing on tensile properties of injection molded biopolyesters based on 2,5-furandicarboxylic acid. Polym Eng Sci. https://doi.org/10.1002/pen.25675
Sousa AF, Vilela C, Fonseca AC et al (2015) Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: a tribute to furan excellency. Polym Chem 6:5961–5983. https://doi.org/10.1039/C5PY00686D
Geng Y, Wang Z, Hu X et al (2019) Bio-based polyesters based on 2,5-furandicarboxylic acid as 3D-printing materials: design, preparation and performances. Eur Polym J 114:476–484. https://doi.org/10.1016/j.eurpolymj.2018.10.041
Papageorgiou GZ, Papageorgiou DG, Terzopoulou Z, Bikiaris DN (2016) Production of bio-based 2,5-furan dicarboxylate polyesters: Recent progress and critical aspects in their synthesis and thermal properties. Eur Polym J 83:202–229. https://doi.org/10.1016/j.eurpolymj.2016.08.004
Jiang M, Liu Q, Zhang Q et al (2012) A series of furan-aromatic polyesters synthesized via direct esterification method based on renewable resources. J Polym Sci Part A Polym Chem 50:1026–1036. https://doi.org/10.1002/pola.25859
Wang J, Liu X, Jia Z et al (2018) Highly crystalline polyesters synthesized from furandicarboxylic acid (FDCA): potential bio-based engineering plastic. Eur Polym J 109:379–390. https://doi.org/10.1016/j.eurpolymj.2018.10.014
Zhang L, Luo X, Qin Y, Li Y (2017) A novel 2,5-furandicarboxylic acid-based bis(cyclic carbonate) for the synthesis of biobased non-isocyanate polyurethanes. RSC Adv 7:37–46. https://doi.org/10.1039/C6RA25045A
Kwiatkowska M, Kowalczyk I, Kwiatkowski K, Zubkiewicz A (2020) Microstructure and mechanical/elastic performance of biobased poly (butylene furanoate)-block-poly (ethylene oxide) copolymers: effect of the flexible segment length. Polym (Basel). https://doi.org/10.3390/polym12020271
Hu H, Zhang R, Sousa A et al (2018) Bio-based poly(butylene 2,5-furandicarboxylate)-b-poly(ethylene glycol) copolymers with adjustable degradation rate and mechanical properties: synthesis and characterization. Eur Polym J 106:42–52. https://doi.org/10.1016/j.eurpolymj.2018.07.007
Kasmi N, Ainali NM, Agapiou E et al (2019) Novel high Tg fully biobased poly(hexamethylene-co-isosorbide-2,5-furan dicarboxylate) copolyesters: Synergistic effect of isosorbide insertion on thermal performance enhancement. Polym Degrad Stab 169:108983. https://doi.org/10.1016/j.polymdegradstab.2019.108983
Paszkiewicz S, Irska I, Zubkiewicz A et al (2021) Biobased thermoplastic elastomers: structure-property relationship of poly(hexamethylene 2,5-furanodicarboxylate)-block-poly(tetrahydrofuran) copolymers prepared by melt polycondensation. Polym (Basel). https://doi.org/10.3390/polym13030397
Terzopoulou Z, Papadopoulos L, Zamboulis A et al (2020) Tuning the properties of furandicarboxylic acid-based polyesters with copolymerization. Polym (Basel) 63(09):594–602. https://doi.org/10.14314/polimery.2018.9.3
Burgess SK, Karvan O, Johnson JR et al (2014) Oxygen sorption and transport in amorphous poly(ethylene furanoate). Polym (Guildf) 55:4748–4756. https://doi.org/10.1016/j.polymer.2014.07.041
Burgess SK, Kriegel RM, Koros WJ (2015) Carbon dioxide sorption and transport in amorphous poly(ethylene furanoate). Macromolecules 48:2184–2193. https://doi.org/10.1021/acs.macromol.5b00333
Zubkiewicz A, Paszkiewicz S, Szymczyk A (2020) Możliwości syntezy poliamidów i kopoliamidów na bazie kwasu 2,5-furanodikarboksylowego oraz jego pochodnych. In: Mołdoch-Mendoń I, Maciąg K (eds) Technologie XXI wieku – aktualne problemy i nowe wyzwania Tom 1. Wydawnictwo Naukowe TYGIEL. Lublin, pp 239–248
Moore JA, Bunting WW (1985) Polyesters and polyamides containing isomeric furan dicarboxylic acids. Polym Sci Technol 31:51–91. https://doi.org/10.1007/978-1-4613-2121-7_3
Luo K, Wang Y, Yu J et al (2016) Semi-bio-based aromatic polyamides from 2,5-furandicarboxylic acid: toward high-performance polymers from renewable resources. RSC Adv 6:87013–87020. https://doi.org/10.1039/c6ra15797a
da Fontoura CM, Pistor V, Mauler RS (2019) Evaluation of degradation of furanic polyamides synthesized with different solvents. Polimeros 29:1–6. https://doi.org/10.1590/0104-1428.08917
Mitiakoudis A, Gandini A (1991) Synthesis and characterization of furanic polyamides. Macromolecules 24:830–835. https://doi.org/10.1021/ma00004a003
Miyagawa N, Suzuki T, Okano K et al (2018) Synthesis of furan dimer-based polyamides with a high melting point. J Polym Sci Part A Polym Chem 56:1516–1519. https://doi.org/10.1002/pola.29031
Fehrenbacher U, Grosshardt O, Kowollik K et al (2009) Synthese und charakterisierung von polyestern und polyamiden auf der basis von furan-2,5-dicarbonsäure. Chemie Ing Tech 81:1829–1835. https://doi.org/10.1002/cite.200900090
Ali DK, Al-Zuheiri AM, Sweileh BA (2020) Green ion-exchange bisfuranic polyamides by polycondensation with bio-based diamines. Green Mater 8:24–31. https://doi.org/10.1680/jgrma.18.00091
Abid S, El Gharbi R, Gandini A (2004) Polyamides incorporating furan moieties. 5. Synthesis and characterisation of furan-aromatic homologues. Polymer (Guildf) 45:5793–5801. https://doi.org/10.1016/j.polymer.2004.06.046
Cureton LST, Napadensky E, Annunziato C, La Scala JJ (2017) The effect of furan molecular units on the glass transition and thermal degradation temperatures of polyamides. J Appl Polym Sci 134:1–12. https://doi.org/10.1002/app.45514
Endah YK, Han SH, Kim JH et al (2016) Solid-state polymerization and characterization of a copolyamide based on adipic acid, 1,4-butanediamine, and 2,5-furandicarboxylic acid. J Appl Polym Sci. https://doi.org/10.1002/app.43391
Cao M, Zhang C, He B et al (2017) Synthesis of 2,5-furandicarboxylic acid-based heat-resistant polyamides under existing industrialization process. Macromol Res 25:722–729. https://doi.org/10.1007/s13233-017-5070-4
Jiang Y, Maniar D, Woortman AJJ et al (2015) Enzymatic polymerization of furan-2,5-dicarboxylic acid-based furanic-aliphatic polyamides as sustainable alternatives to polyphthalamides. Biomacromol 16:3674–3685. https://doi.org/10.1021/acs.biomac.5b01172
Jiang Y, Maniar D, Woortman AJJ, Loos K (2016) Enzymatic synthesis of 2,5-furandicarboxylic acid-based semi-aromatic polyamides: enzymatic polymerization kinetics, effect of diamine chain length and thermal properties. RSC Adv 6:67941–67953. https://doi.org/10.1039/C6RA14585J
Huang W, Zhai J, Zhang C et al (2020) 100% Bio-based polyamide with temperature/ultrasound dually triggered reversible cross-linking. Ind Eng Chem Res 59:13588–13594. https://doi.org/10.1021/acs.iecr.0c02028
Cousin T, Galy J, Rousseau A, Dupuy J (2018) Synthesis and properties of polyamides from 2,5-furandicarboxylic acid. J Appl Polym Sci 135:45901. https://doi.org/10.1002/app.45901
Triki R, Abid M, Tessier M et al (2013) Furan-based poly(esteramide)s by bulk copolycondensation. Eur Polym J. https://doi.org/10.1016/j.eurpolymj.2013.04.014
Wilsens CHRM, Deshmukh YS, Noordover BAJ, Rastogi S (2014) Influence of the 2, 5-furandicarboxamide moiety on hydrogen bonding in aliphatic-aromatic poly(ester amide)s. Macromolecules 47:6196–6206. https://doi.org/10.1021/ma501310f
Maniar D, Silvianti F, Ospina VM et al (2020) On the way to greener furanic-aliphatic poly(ester amide)s: enzymatic polymerization in ionic liquid. Polymer (Guildf) 205:122662. https://doi.org/10.1016/j.polymer.2020.122662
Kluge M, Papadopoulos LM, Tzetzis A, Zamboulis D, Alexandra Bikiaris DN, Robert T (2020) A facile method to synthesize semicrystalline poly(ester amide)s from 2,5-furandicarboxylic acid, 1,10-decanediol, and crystallizable amido diols. CS Sustain Chem Eng 8:10812–10821. https://doi.org/10.1021/acssuschemeng.0c02730
Elamri A, Zdiri K, Harzallah O, Lallam A (2017) Progress in Polyethylene Terephthalate Recycling. Uses Properties and Degradation. Nova Science Publishers, Polyethylene Terephthalate, p 33
Paszkiewicz S, Janowska I, Pawlikowska D et al (2018) New functional nanocomposites based on poly(trimethylene 2,5-furanoate) and few layer graphene prepared by in situ polymerization. Express Polym Lett 12:530–542. https://doi.org/10.3144/expresspolymlett.2018.44
Paszkiewicz S, Irska I, Piesowicz E (2020) Environmentally friendly polymer blends based on post-consumer glycol-modified poly(ethylene terephthalate) (PET-G) foils and poly(Ethylene 2,5-Furanoate) (PEF): preparation and characterization. Materials (Basel) 13:2673. https://doi.org/10.3390/ma13122673
Abdollahi H, Salimi A, Barikani M et al (2019) Systematic investigation of mechanical properties and fracture toughness of epoxy networks: role of the polyetheramine structural parameters. J Appl Polym Sci 136:47121. https://doi.org/10.1002/app.47121
Gomes M, Gandini A, Silvestre AJD, Reis B (2011) Synthesis and characterization of poly(2,5-furan dicarboxylate)s based on a variety of diols. J Polym Sci Part A Polym Chem 49:3759–3768. https://doi.org/10.1002/pola.24812
Abid M, El Gharbi R, Gandini A (2000) Polyamides incorporating furan moieties. 3. Polycondensation of 2- furamide with paraformaldehyde. Polymer (Guildf) 41:3555–3560. https://doi.org/10.1016/S0032-3861(99)00538-8
Sanusi OM, Papadopoulos L, Klonos PA et al (2020) Calorimetric and dielectric study of renewable poly(hexylene 2,5-furan-dicarboxylate)-based nanocomposites in situ filled with small amounts of graphene platelets and silica nanoparticles. Polymers (Basel) 12:1239. https://doi.org/10.3390/polym12061239
Papageorgiou GZ, Tsanaktsis V, Papageorgiou DG et al (2015) Furan-based polyesters from renewable resources: crystallization and thermal degradation behavior of poly(hexamethylene 2,5-furan-dicarboxylate). Eur Polym J 67:383–396. https://doi.org/10.1016/j.eurpolymj.2014.08.031
Burgess SK, Leisen JE, Kraftschik BE et al (2014) Chain mobility, thermal, and mechanical properties of poly(ethylene furanoate) compared to poly(ethylene terephthalate). Macromolecules 47:1383–1391. https://doi.org/10.1021/ma5000199
Kwiatkowska M, Kowalczyk I, Szymczyk A, Gorący K (2018) Effect of thermal aging on the crystalline structure and mechanical performance of fully bio-based, furan-ester, multiblock copolymers. Polimery 63(09):594–602
Genovese L, Soccio M, Lotti N et al (2018) Effect of chemical structure on the subglass relaxation dynamics of biobased polyesters as revealed by dielectric spectroscopy: 2,5-furandicarboxylic acid vs. trans -1,4-cyclohexanedicarboxylic acid. Phys Chem Chem Phys 20:15696–15706. https://doi.org/10.1039/c8cp01810c
Thiyagarajan S, Vogelzang W, Knoop JI et al (2014) Biobased furandicarboxylic acids (FDCAs): effects of isomeric substitution on polyester synthesis and properties. Green Chem 16:1957–1966. https://doi.org/10.1039/C3GC42184H
Papageorgiou GZ, Papageorgiou DG, Tsanaktsis V, Bikiaris DN (2015) Synthesis of the bio-based polyester poly(propylene 2,5-furan dicarboxylate). Comparison of thermal behavior and solid state structure with its terephthalate and naphthalate homologues. Polymer (Guildf) 62:28–38. https://doi.org/10.1016/j.polymer.2015.01.080
Gao H, Bai Y, Liu H, He J (2019) Mechanical and gas barrier properties of structurally enhanced poly(ethylene terephthalate) by introducing 1,6-hexylenediamine unit. Ind Eng Chem Res 58:21872–21880. https://doi.org/10.1021/acs.iecr.9b04953
Yang Z-Y, Chou Y-L, Yang H-C et al (2021) Synthesis and characterization of thermoplastic poly(Ester Amide)s elastomer (TPEaE) obtained from recycled PET. J Renew Mater 9:867–880. https://doi.org/10.32604/jrm.2021.014476
Wang X, Liu S, Wang Q et al (2018) Synthesis and characterization of poly(ethylene 2,5-furandicarboxylate-co-ε-caprolactone) copolyesters. Eur Polym J 109:191–197. https://doi.org/10.1016/j.eurpolymj.2018.09.051
Funding
The studies were financed by the National Science Centre within project SONATA no 2018/31/D/ST8/00792.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Gregory Rutledge.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zubkiewicz, A., Irska, I., Miadlicki, P. et al. Structure, thermal and mechanical properties of copoly(ester amide)s based on 2,5‐furandicarboxylic acid. J Mater Sci 56, 19296–19309 (2021). https://doi.org/10.1007/s10853-021-06576-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06576-9