
Abstract
Porphyromonas gingivalis (P. gingivalis) is an important pathogen that contributes to periodontal disease and causes infections that promote the progression of atherosclerosis. Our previous studies showed that macrophage migration inhibitory factor (MIF) facilitates monocyte adhesion to endothelial cells by regulating the expression of intercellular adhesion molecule-1 (ICAM-1) inP. gingivalis-infected endothelial cells. However, the detailed pathological role of MIF has yet to be elucidated in this context. To explore the functional receptor(s) of MIF that underlie its participation in the pathogenesis of atherosclerosis, we investigated the expression of the chemokine receptors CD74 and CXCR4 in endothelial cells, both of which were shown to be involved in the adhesion of monocytes to endothelial cells pretreated with P. gingivalis. Furthermore, the formation of a MIF, CD74, and CXCR4 ligand-receptor complex was revealed by our immunofluorescence staining and coimmunoprecipitation results. By interacting with the CD74/CXCR4 receptor complex, MIF may act as a crucial regulator of monocyte-endothelial cell adhesion and promote the atherosclerotic plaque formation induced by P. gingivalis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Periodontitis is one of the most prevalent infectious diseases in the human oral cavity. As a major periodontal pathogen, the presence of Porphyromonas gingivalis (P. gingivalis) in atherosclerotic lesions suggests an association between periodontitis and atherosclerosis [1, 2], a relationship that has been confirmed by epidemiological data, clinical studies, and animal experiments [3,4,5,6,7]. In addition,in vitro research has shown that P. gingivalis can increase the expression of cell adhesion molecules, proinflammatory cytokines, and chemokines in endothelial cells, which have crucial roles in the recruitment of monocytes to the vascular endothelium and the subsequent formation of atherosclerotic plaques [8,9,10,11,12].
The recruitment and adhesion of monocytes via the synergistic responses of multiple chemokines and their receptors has been shown to be crucial events underlying atherosclerotic lesion formation and disease progression [13, 14]. As a highly conserved and atypical proinflammatory cytokine with chemokine-like functions, macrophage migration inhibitory factor (MIF) exerts multipotent immune functions in chronic inflammatory diseases, such as rheumatoid arthritis, atherogenesis, and cancer [15,16,17]. MIF has been demonstrated to primarily promote atherosclerosis through the enhancement of macrophage and T cell recruitment by directly affecting endothelial-monocyte interactions [18, 19].
We previously reported that P. gingivalis infections enhance endothelial MIF and intercellular adhesion molecule-1 (ICAM-1) expression, in addition to promoting the adhesion of monocytes to endothelial cells [20]. Furthermore, we demonstrated that the increased adhesive properties induced byP. gingivalis were dependent on MIF expression [21]. Our findings suggested that P. gingivalis infections lead to endothelial activation and pro-atherosclerotic lesion formation. During this inflammatory process, MIF may undertake a regulator role in monocyte recruitment and atherogenesis.
MIF mediates cellular responses and triggers several signaling pathways by binding to its receptors [22, 23]. Although advances have recently been made in understanding how P. gingivalis promotes atherosclerosis [2, 24], a detailed understanding of how the activities of MIF and its functional receptors participate in atherosclerotic diseases remains unclear. In this study, we investigated potential MIF receptors that facilitate ICAM-1 expression and monocyte adhesion to endothelial cells to provide new insights into the pathogenesis of P. gingivalis-promoted atherosclerosis. The results of our study revealed the molecular mechanism of MIF regulation of monocyte-endothelial cell adhesion. We demonstrated that MIF is a functional ligand of chemokine receptors CD74 and CXCR4 and that it participates in the regulation of monocyte recruitment in atherosclerosis promoted by P. gingivalis infection.
MATERIALS AND METHODS
Cells
EA.hy926 cells (a human umbilical vein endothelial cell line) and THP-1 cells (a monocyte cell line) were used in our study, both of which were acquired from Keygen Biotech Company (Nanjing, China). EA.hy926 cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Gibco BRL, Carlsbad, CA, USA) supplemented with 15% fetal bovine serum (FBS; GeneTimes, Shanghai, China), and THP-1 cells were grown in DMEM containing 10% FBS. Both cell lines were cultured at 37 °C with 5% CO2. A trypan blue exclusion test was used to assess cell viability. The EA.hy926 cells were used in the following assays when the observed cell viability was > 90%. Before the two cell lines were co-cultured, the fluorescent dye calcein-AM (0.1 mg/mL; BioVision, Bay Area, CA, USA) was used to label the THP-1 cells in the dark for 30 min.
Bacterial Strain
P. gingivalis ATCC 33277 was routinely maintained in brain heart infusion broth supplemented with 5% defibrinated sheep’s blood, 0.5% yeast, 0.1% menadione, and 1% hemin and was cultured under anaerobic conditions (80% N2, 10% O2, and 10% H2) at 37 °C. The bacterial cells were collected, and the optical density of the bacterial suspension was adjusted to 1.0 at 600 nm before infecting EA.hy926 cells.
Analysis of CD74 and CXCR4 Expression by Western Blot
EA.hy926 cells were infected with P. gingivalis at a multiplicity of infection (MOI) of 100 for 24 h, after which the expression of CD74 and CXCR4 was assessed by Western blot. Cells cultured without P. gingivalis were used as a negative control.
After the cells were lysed, the protein concentration in cell lysates was determined by a BCA assay. The samples were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane, with GAPDH used as a loading control. After blocking, proteins of interest were detected with specific primary antibodies, including a mouse anti-CD74 mAb (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), a mouse anti-CXCR4 mAb (1:500; Proteintech, Rosemont, IL, USA), and a mouse anti-GAPDH antibody (1:1000; Wanlei, Shenyang, China). After an overnight incubation, the blots were washed and then incubated with Dylight 800 conjugated rabbit anti-mouse IgG (1:1000; Abbkine, Inc., Redlands, CA, USA) for 1 h. Odyssey CLX (LI-COR, Lincoln, NE, USA) was exploited for Western blot analyses. The relative protein expression levels were presented.
Analysis of ICAM-1 Protein and Gene Transcription by Western Blot and qRT-PCR
Endothelial cells were pretreated with a neutralizing antibody of CD74 (C-16, 5 μg/mL; Santa Cruz Biotechnology) [22, 25], an inhibitor of CXCR4 (AMD3100, 20 nM; Abcam, Cambridge, MA, UK) [22, 25] or DMEM medium for 1 h. Next, the cells were infected byP. gingivalis for 24 h (MOI = 100). The cells treated with culture medium only were used as a control. Then, the whole cell protein was extracted and samples were analyzed for ICAM-1 expression by Western blot as described above using rabbit anti-ICAM-1 mAb (1:500; Wanlei, Shenyang, China) and Dylight 800 conjugated goat anti-rabbit IgG (1:1000; Abbkine, Inc.) antibodies.
Using cells that were treated as described above, a quantitative real-time polymerase chain reaction (qRT-PCR) assay was performed as described in our previous study [21]. Briefly, TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was utilized to extract total cellular RNA, the purity of which was evaluated by determining the 260/280 nm absorbance ratio. Biosystems 7500 Fast Real-Time PCR System (RR047, RR420, Takara, Tokyo, Japan) was used to analyze the ICAM-1 mRNA expression, together with the SYBR® Premix Ex Taq™ II (RR047, RR420, Takara, Tokyo, Japan), which was used according to the manufacturer’s protocol. The following primers were used for qRT-PCR: ICAM-1 forward: 5′-TGATGGGCAGTCAACAGCTA-3′, ICAM-1 reverse: 5′-GCGTAGGGTAAGGTTCTTGC-3′, GAPDH forward: 5′-GAAGGTCGGAGTCAACGGAT-3′, GAPDH reverse: 5′-CCTGGAAGATGGTGATGGGAT-3′. The primers of ICAM-1 and GAPDH were designed by Primer 3, and the specificity was verified by blasting primer sequences against the NCBI database. The mRNA level of the internal reference GAPDH was designed as 100%, and ICAM-1 mRNA is presented compared to the GAPDH reference.
Adhesion Assays of THP-1 to EA.hy926 Cells
Endothelial cells were grown on six-well plates until a confluent monolayer was formed, after which the cells were treated as described above. Later, THP-1 cells (1 × 106, labeled with 5 μM calcein-AM) were co-cultured with endothelial cells for an additional 1 h at 37 °C with 5% CO2 keeping in the dark. Next, the non-adherent THP-1 cells were rinsed with PBS. After being fixed with a 4% formaldehyde solution, the labeled monocytes adhered to the surface of EA.hy926 cells were examined with a fluorescence microscope (Nikon 80i, Tokyo, Japan). Three fields were chosen randomly and the adherent monocytes were identified through visual cell counting.
Immunofluorescence Staining and Colocalization Analysis
Endothelial cells were cultured on cover slips in 24-well plates at a concentration 4 × 104 cells per well for 24 h at 37 °C. Next, the cells were infected with P. gingivalis at an MOI of 100 for 24 h. The cells were subsequently washed and fixed, after which they were incubated in 1% BSA in 0.1% PBS-Tween for 1 h. Afterwards, the samples were incubated with mouse anti-CD74 (1:100; Santa Cruz) and rabbit anti-CXCR4 (1:200; Abcam) overnight at 4 °C. Donkey anti-mouse IgG labeled with PE (1:20; Proteintech) and goat anti-rabbit IgG labeled with FITC (1:20; Proteintech) were used as the secondary antibodies and were incubated with the cells for 1 h at room temperature. DAPI (Boster, Wuhan, China) was used to stain the cell nucleus (pseudo-colored blue), and the cells were observed using a fluorescence microscope (Nikon 80i, Japan) to evaluate the colocalization between CXCR4 and CD74.
Transfection Assays of EA.hy926 Cells
The plasmid pGCsi-H1 Neo GFP (Genechem, Shanghai, China) was used to construct a stably transfected EA.hy926 cell line in which CXCR4 was silenced. shRNA oligonucleotide fragment interference vectors with interference segment information 5′-GGGUGUGAGUUUGAGAACA-3′ were established, and cells transfected with empty vectors were used as a negative control. EA.hy926 cells in logarithmic growth phase were transfected using lipofectamine 2000 (Invitrogen) as a transfection reagent. Pure cell lines were obtained by selection with G418 (500 μg/ml; Invitrogen). The cells were incubated for 48 h, and the total cellular CXCR4 content was determined by anti-CXCR4 Western blot and qRT-PCR to assess the knock down efficiency. The positive monoclonal cell line was cultured in DMEM containing 15% FBS at 37 °C with 5% CO2 and was used in subsequent assays.
Coimmunoprecipitations and Pull-Down Experiments
EA.hy926 cells stably transfected with the CXCR4-silencing plasmid, normal EA.hy926 cells, and the negative control transfected cells were infected with P. gingivalis at an MOI of 100 for 24 h. Next, whole cell protein was extracted from the samples. After diluting the cell proteins with PBS to 1 μg/μl, samples were incubated with a rabbit anti-MIF mAb (1 μl/500 μl; Abcam) or an IgG control overnight at 4 °C for immunoprecipitation. Protein A agarose beads were washed twice with PBS and then were diluted to a concentration of 50% by PBS. Next, the prepared protein-A agarose beads and the lysates were incubated together for 2 h at 4 °C, after which the beads bound with the immune complexes were washed three times with pre-chilled PBS. The beads were subsequently resuspended in sample buffer and boiled, and then the boiled samples subsequently used for Western blot to assess the presence of MIF, CXCR4, and CD74. Blots were obtained by using rabbit anti-MIF mAb (1:1000; Abcam), mouse anti-CD74 mAb (1:500; Santa Cruz), and mouse anti-CXCR4 mAb (1:500; Proteintech). To ascertain uniformity of protein loading, the membrane was immunoblotted with anti-GAPDH (1:1000; Wanlei). Goat anti-rabbit or rabbit anti-mouse Dylight 800 conjugated IgG (1:1000; Abbkine, Inc.) were used as secondary antibodies and were incubated with the blots for 1 h at room temperature. Lysates without immunoprecipitation were tested by Western blot as controls.
Statistical Analysis
Data are presented as the means ± SD of three repeated experiments. Student’s t test in SPSS 17.0 was used to evaluate the significant differences between the groups. Differences with aP < 0.05 were considered significant.
RESULTS
P. gingivalis Infection Induces the Expression of CXCR4 but Not CD74 in Endothelial Cells
We previously reported that P. gingivalis infection facilitates the expression of ICAM-1 and the adhesion of THP-1 and EA.hy926 cells by upregulating the secretion of MIF [21]. These findings prompted us to investigate the mechanisms by which MIF promotes the adhesion of the endothelial cells, which are closely associated with the inflammatory process of atherosclerosis.
To evaluate the involvement of possible MIF receptors in this process, we examined the expression of CD74 and CXCR4 in EA.hy926 cells. The Western blot results showed that the expression of CXCR4 but not CD74 in EA.hy926 cells was induced by P. gingivalis. Compared to the un-infected control cells, CXCR4 expression was increased 1.75-fold by P. gingivalis infection (24 h, MOI = 100; P < 0.01). In contrast, CD74 expression remained unchanged after P. gingivalis infection (Fig. 1).

P. gingivalis infection induced the protein level of CXCR4 but not CD74 in EA.hy926 cells. P. gingivalis infected EA.hy926 cells for 24 h at MOI = 100, then the expression of CD74 and CXCR4 was detected by Western blot. Cells cultured without P. gingivalis were used as a control. a Western blot analysis of CD74 and CXCR4 in endothelial cells. b Quantitative analysis of the Western blot. Data were presented compared to the GAPDH reference. *P < 0.01.
P. gingivalis Induction of ICAM-1 andICAM-1 mRNA Expression Is Partially Dependent on CD74 and CXCR4
P. gingivalis infection was previously shown to enhance ICAM-1 mRNA and protein expression in endothelial cells. In this study, we explored the roles of the receptors CD74 and CXCR4 on changes in ICAM-1 levels. We analyzed the expression of ICAM-1 after specifically blocking CD74 or CXCR4. A neutralizing CD74 antibody (C-16) and a CXCR4 inhibitor (AMD3100) were individually added to EA.hy926 cells for 1 h before being treated with P. gingivalis. Our results demonstrated that the expression of ICAM-1 was significantly upregulated by P. gingivalis (Fig. 2a–c), and this induction by P. gingivalis was counteracted by the neutralizing antibody of CD74 or the CXCR4 inhibitor treatments. Compared with the cells infected by P. gingivalis alone, C-16 or AMD3100 reduced the ICAM-1 protein level by 38% (P < 0.01) and 50% (P < 0.01), respectively (Fig. 2a, b). Further confirmation of these results was obtained through our qRT-PCR findings. The expression profile of ICAM-1 mRNA was consistent with that of ICAM-1 protein levels, as ICAM-1 mRNA levels were reduced by 66% (P < 0.01) and 82% (P < 0.01) by the addition of C-16 or AMD3100, respectively (Fig. 2c). It was notified that the ICAM-1 mRNA level in the AMD3100 treatment group was lower than the control. But the statistical analysis showed there was no significant difference between the two groups. Our results indicated that the induction of ICAM-1 and ICAM-1 mRNA in endothelial cells by P. gingivalis was partially dependent on CD74 and CXCR4.

P. gingivalis induction of ICAM-1 and ICAM-1 mRNA expression is partially dependent on CD74 and CXCR4. EA.hy926 cells were infected with P. gingivalis at MOI = 100 for 24 h after the addition of C-16 or AMD3100, then the level of ICAM-1 and ICAM-1 mRNA was determined by Western blot and qRT-PCR. EA.hy926 cells cultured in medium only were used as a negative control. a Western blot analysis of ICAM-1.b Quantitative analysis of Western blot. c Quantitative real-time PCR analysis of ICAM-1 mRNA. *P < 0.01.
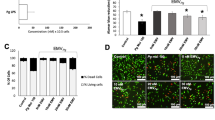
Enhanced Adhesion of Monocytes to P. gingivalis-Infected Endothelial Cells Is Partially Dependent on CD74 and CXCR4
Our early assays indicated that the enhanced adhesion of monocytes to endothelial cells induced by P. gingivalis was regulated by MIF. In this assay, we evaluated the possible roles of the receptors CD74 and CXCR4 in the observed monocyte-endothelial cell adhesion. Bacterial infection has been previously observed to increase cell adhesion by 8.17-fold [21]. Compared to the endothelial cells infected with P. gingivalis, the C-16 and AMD3100 pretreatments reduced monocyte adhesion by 54% (P < 0.01) and 41%, respectively (P < 0.01; Fig. 3). Thus, the results of this assay showed that the P. gingivalis-induced promotion of monocyte-endothelial cell adhesion is also dependent on CD74 and CXCR4.

Enhanced THP-1 cells adhesion to P. gingivalis-infected EA.hy926 cells is partially dependent on CD 74 and CXCR4. The EA.hy926 cells were pre-incubated with C-16 (5 μg/mL) or AMD3100 (20 nM) for 1 h then infected with P. gingivalis for 24 h (MOI = 100). THP-1 cells labeled with Calcein-AM (5 μM) were co-cultured with EA.hy926 cells for additional 1 h before the adhesion assay. The control group was EA.hy926 cells pre-treated with culture medium only.a Calcein-AM labeled THP-1 cells adhered to EA.hy926 cells under fluorescence microscope (upper) or microscope (lower) (magnification × 100). Representative pictures were captured in three independent experiments. b Cell count assay to evaluate the adherent THP-1 cells. *P < 0.01. Scale bar = 100 μm.
CD74 and CXCR4 Colocalize in EA.hy926 Cells Infected with P. gingivalis
CD74 and CXCR4 have been reported to form a heteromeric receptor complex involved in MIF endocytosis, and colocalization of CD74 and CXCR4 has been observed [26]. Based on our results described above, we were interested in the possibility of the involvement of CD74 and CXCR4 colocalization in MIF regulation of monocyte adhesion to endothelial cells. Here, we observed both CD74 and CXCR4 by fluorescence microscopy. The results of immunofluorescence staining indicated that there was no significant enhancement in the surface expression of CD74. In contrast to CD74, the expression of CXCR4 was increased significantly byP. gingivalis. The results of IF staining showed the colocalization of CD74 and CXCR4 (Fig. 4).

Colocalization of CD74 and CXCR4 in EA.hy926 cells infected with P. gingivalis. EA.hy926 cells infected with P. gingivalis (24 h, MOI = 100) were observed by fluorescence microscopy (bottom). The cells cultured with medium alone were used as a control (top). Colocalization of CD74 and CXCR4 in the plasma membrane (orange-yellow overlay) was shown. After three independent experiments, representative pictures were captured by fluorescence microscope (magnification × 400). Scale bar = 20 μm.
Receptor Complex Formation Between CD74 and CXCR4 for MIF in EA.hy926 Cells Infected with P. gingivalis
To verify whether CD74 and CXCR4 bind together to form a receptor complex in endothelial cells infected with P. gingivalis, coimmunoprecipitation and pull-down assays were performed. When lysates of EA.hy926 cells infected with P. gingivalis were immunoprecipitated with an anti-MIF antibody, a subsequent Western blot assay showed that CD74 and CXCR4 coprecipitated. In contrast, when lysates of endothelial cells stably transfected with theCXCR4-silencing plasmid were immunoprecipitated with the anti-MIF antibody, the levels of CD74, CXCR4, and MIF proteins were markedly decreased compared to the corresponding control (Fig. 5). Thus, in line with the suggestion that the level of ICAM-1 in endothelial cells and monocyte-endothelial cell adhesion induced by P. gingivalis infection is regulated by MIF, we present the hypothesis that portions of CD74 and CXCR4 may bind to form a receptor complex for MIF in endothelial cells infected with P. gingivalis. This interaction was further confirmed by the results of the fluorescence colocalization analysis and coimmunoprecipitations/pull-down experiments.

Receptor complex formation between CD74 and CXCR4 for MIF in EA.hy926 cells infected with P. gingivalis. Input controls (lysates without immunoprecipitation) are shown (a). Coimmunoprecipitation and pull-down assays were performed by anti-MIF antibody (b) or anti-IgG antibody (c) for immunoprecipitation (IP), and then anti-MIF antibody, anti-CXCR4 antibody, and anti-CD74 antibody were used for Western blot (WB). A: EA.hy926 cells infected withP. gingivalis for 24 h (MOI = 100). B: EA.hy926 cells stably transfected with empty vectors and infected with P. gingivalis. C: EA.hy926 cells stably transfected with CXCR4-silencing plasmid and infected with P. gingivalis. Data were representative of three independent experiments.
DISCUSSION
MIF has been demonstrated to be closely associated with the progression and severity of atherosclerosis [17]. Animal experiments have shown that MIF is correlated with the thickening of the aortic intima and lipid deposition in mice and in rabbits fed an atherogenic diet [27, 28]. In contrast, Mif blockade in mice results in a regression of plaque areas [29]. Furthermore, antibody inhibition and genetic deletion studies have revealed that MIF influences the promotion of atherosclerosis by enhancing macrophage and T cell recruitment [21, 30].
P. gingivalis is considered to be a significant pathogen of periodontal disease and has been demonstrated to participate in the development of atherosclerosis. Interestingly, P. gingivalis DNA has been detected in atheromatous plaques [6, 31, 32]. Experiments in low-density lipoprotein (Ldlp)- and apolipoprotein E (Apoe)-deficient mice showed that P. gingivalis infection promotes atherosclerosis by markedly increasing lesion size and disease progression, which is followed by endothelial function impairment and systemic inflammation [3, 33]. Our early in vitro studies ascertained that P. gingivalis infection enhances the level of ICAM-1 in EA.hy926 cells and THP-1-EA.hy926 cell adhesion, demonstrating that P. gingivalis contributes to pro-atherosclerotic changes in endothelial cells [20]. Furthermore, we observed that P. gingivalis infection promoted MIF secretion in endothelial cells and that MIF was involved in the atherosclerotic plaque formation induced by P. gingivalis [21]. Currently, our understanding of the mechanisms by whichP. gingivalis facilitates endothelial adhesion molecule expression and monocyte-endothelial cell adhesion is limited.
The functional receptor(s) of MIF and the molecular modes underlying its role in inflammatory diseases have remained elusive for decades. Recently, as a result of identifying CD74, CXCR4, and CXCR2 as receptors for MIF, we have gained a better understanding of the molecular mechanisms involved in MIF-mediated signaling pathways [29]. Depending on the cell type and its associated receptor expression profile, the activation of MIF is mediated by the different receptors [22, 25]. CD74 is a protein that participates in the formation and transport of MHC class II proteins and lacks signal-transducing intracellular domain [29, 34,35,36]. MIF binds to the extracellular domain of CD74 to form a ligand-receptor complex that activates multiple signaling pathways to participate in inflammatory responses [34, 35]. CXCR2 and CXCR4 may serve as additional signal transduction receptors of CD74 in MIF-stimulated inflammatory response [22, 29]. CXCR2 and CXCR4 are CXC chemokine receptors, and both of them belong to G protein-coupled receptor family [37, 38]. The colocalization of CXCR2 and CD74 in the cell membrane has been observed [23, 29]. The interaction between CXCR2 and CD74 suggests that MIF may affect downstream signal transduction through functional CXCR/CD74 complexes. Simons et al. found that MIF-mediated chemotaxis of endothelial progenitor cells through CXCR4 [39]. It was observed that MIF activates JNK signaling and upstream kinases PI3K and Src by binding to CXCR4 and CD74, thereby regulating inflammation, cell differentiation, and apoptosis [22]. Schwartz et al. confirmed that CXCR4 and CD74 colocalize in the cell membrane, forming a functional receptor complex to mediate MIF-specific signal transduction [26, 40]. Thus, this evidence indicates that functional CD74/CXCR receptor complexes may occur. However, in the presence of P. gingivalis, the specific receptor(s) or receptor complex of MIF have not yet been defined.
In the current study, we focused on the combination of CD74, CXCR4, and MIF. By analyzing the receptor protein expression, we showed that the expression of CXCR4 was significantly enhanced in endothelial cells infected with P. gingivalis, independent on CD74 co-expression. In contrast, both CD74 and CXCR4 were shown to be involved in the increased ICAM-1 levels in EAhy.926 cells and in the observed enhanced THP-1-EAhy.926 cell adhesion. Thus, we present the hypothesis that CXCR4 and CD74 form a receptor complex that is responsive to MIF. First, through fluorescence microscopy observations, we demonstrated that CD74 and CXCR4 colocalized at the surfaces of infected endothelial cells. Subsequently, we investigated whether CD74 interacts with CXCR4 in endothelial cells infected with P. gingivalis. The co-immunoprecipitation and pull-down analyses were performed using stably transfected EA.hy926 cells with the CXCR4-silencing plasmid and an MIF antibody. Our results provided a hint to the functional interplay between CD74 and CXCR4, revealing that CD74 and CXCR4 form a receptor complex that bind to MIF in infected EA.hy926 cells. We speculate that MIF activates monocyte adhesion to the vascular endothelial cell surface, promoting plaque formation associated with atherosclerotic disease by interacting with CD74 and CXCR4. Furthermore, it is conceivable that CD74/CXCR4 complexes play a crucial part in P. gingivalis-induced atherogenic lesion formation. Interestingly, we also found that when the CXCR4 gene was knocked down, it seemed that the expression of MIF decreased to some extent. We hypothesized that due to the redundant unbound MIF by CXCR4 gene silence, some uncertain negative feedback might be activated, resulting in the inhibition of MIF synthesis. This potential association between MIF and CXCR4 requires our further experiments to confirm.
Overall, the results of our study indicated that the CD74/CXCR4 complex was responsive to MIF target cell activation upon P. gingivalis infection. However, it has been reported that CD74 can also be combined with other proteins, such as CXCR2 in signal transduction of MIF. In addition, it is suggested that blocking the signal transduction of MIF can be used as a therapeutic target in inflammatory diseases, such as atherosclerosis. Considering the multiple receptors of MIF, we believe that a combination of blocking each receptor may achieve better therapeutic effects. Therefore, additional experiments are needed to identify other proteins bound to CD74 or CXCR4 during theP. gingivalis infection process.
CONCLUSION
Our experiments revealed that CD74 and CXCR4 are involved in the pathological processes by which P. gingivalis promotes ICAM-1 expression and monocyte-endothelial cell adhesion. MIF-CD74-CXCR4 ligand-receptor complexes were present in endothelial cells infected with P. gingivalis, which may play a significant role inP. gingivalis-induced atherosclerosis.
Abbreviations
- P. gingivalis :
-
Porpyromonas gingivalis
- MIF:
-
macrophage migration inhibitory factor
- ICAM-1:
-
intercellular adhesion molecule-1
- DMEM:
-
Dulbecco’s modified Eagle medium
- FBS:
-
fetal bovine serum
- MOI:
-
multiplicity of infection
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
References
Rocco, C.J., L.O. Bakaletz, and S.D. Goodman. 2018. Targeting the HUβ Protein Prevents Porphyromonas gingivalis from Entering into Preexisting Biofilms. Journal of Bacteriology 200: e00790–e00717.
Hussain, M., C.M. Stover, and A. Dupont. 2015. P. gingivalis in Periodontal Disease and Atherosclerosis—Scenes of Action for Antimicrobial Peptides and Complement.Frontiers in Immunology 6: 45.
Chistiakov, D.A., A.N. Orekhov, and Y.V. Bobryshev. 2016. Links between atherosclerotic and periodontal disease. Experimental and Molecular Pathology 100: 220–235.
Zhang, B., H. Khalaf, A. Sirsjo, and T. Bengtsson. 2015. Gingipains from the Periodontal Pathogen Porphyromonas gingivalis play a Significant Role in Regulation of Angiopoietin 1 and Angiopoietin 2 in Human Aortic Smooth Muscle Cells. Infection and Immunity 83: 4256–4265.
Li, C., Z. Lv, Z. Shi, Y. Zhu, Y. Wu, L. Li, and Z. Iheozor-Ejiofor. 2014. Periodontal therapy for the management of cardiovascular disease in patients with chronic periodontitis. Cochrane Database Syst ematic Reviews 8: CD009197.
Mougeot, J.C., C.B. Stevens, B.J. Paster, M.T. Brennan, P.B. Lockhart, and F.K. Mougeot. 2017. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. Journal of Oral Microbiology 9: 1281562.
Al Batran, R., F. Al-Bayaty, M.M. Al-Obaidi, and A. Ashrafi. 2014. Insights into the antiatherogenic molecular mechanisms of andrographolide against Porphyromonas gingivalis-induced atherosclerosis in rabbits. Naunyn Schmiedebergs Archives of Pharmacology 387: 1141–1152.
Hashizume, T., T. Kurita-Ochiai, and M. Yamamoto. 2011. Porphyromonas gingivalis stimulates monocyte adhesion to human umbilical vein endothelial cells. FEMS Immunology and Medical. Microbiology 62: 57–65.
Roth, G.A., B. Moser, F. Roth-Walter, M.B. Giacona, E. Harja, P.N. Papapanou, A.M. Schmidt, and E. Lalla. 2007. Infection with a periodontal pathogen increases mononuclear cell adhesion to human aortic endothelial cells.Atherosclerosis 190: 271–281.
Wan, M., J. Liu, and X. Ouyang. 2015. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappa B pathway. Journal of Periodontal Research 50: 189–196.
Liu, B., J. Wang, L. Cheng, and J. Liang. 2013. Role of JNK and NF-κB pathways in Porphyromonas gingivalis LPS-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. Molecular Medicine Reports 8: 1594–1600.
Libby, P. 2002. Inflammation in atherosclerosis. Nature 420: 868–874.
Weber, C., and H. Noels. 2011. Atherosclerosis: current pathogenesis and therapeutic options. Nature Medicine 17: 1410–1422.
Herbin, O., A.G. Regelmann, B. Ramkhelawon, E.G. Weinstein, K.J. Moore, and K. Alexandropoulos. 2016. Monocyte Adhesion and Plaque Recruitment During Atherosclerosis Development Is Regulated by the Adapter Protein Chat-H/SHEP1. Arteriosclerosis Thrombosis and Vascular Biology 36: 1791–1801.
Morand, E.F., M. Leech, and J. Bernhagen. 2006. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nature Reviews Drug Discovery 5: 399–410.
Kok, T., A.A. Wasiel, R.H. Cool, B.N. Melgert, G.J. Poelarends, and F.J. Dekker. 2018. Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) as an emerging class of therapeutics for immune disorders.Drug Discovery Today 23: 1910–1918.
Tilstam, P.V., D. Qi, L. Leng, L. Young, and R. Bucala. 2017. MIF family cytokines in cardiovascular diseases and prospects for precision-based therapeutics. Expert Opinion on Therapeutic Targets 21: 671–683.
van der Vorst, E.P., Y. Doring, and C. Weber. 2015. Chemokines and their receptors in Atherosclerosis. Journal of Molecular Medicine (Berlin, Germany) 93: 963–971.
Chen, D., M. Xia, C. Hayford, el-L. Tham, V. Semik, S. Hurst, V. Semik, S. Hurst, Y. Chen, H.H. Tam, J. Pan, Y. Wang, X. Tan, H.Y. Lan, H. Shen, V.V. Kakkar, Q. Xu, J.H. McVey, and A. Dorling. 2015. Expression of Human Tissue Factor Pathway Inhibitor on Vascular Smooth Muscle Cells Inhibits Secretion of Macrophage Migration Inhibitory Factor and Attenuates Atherosclerosis in ApoE (-/-) Mice. Circulation 131: 1350–1360.
Zhang, D., H. Zheng, J. Zhao, L. Lin, C. Li, and J. Liu. 2011.Porphorymonas gingivitis induces intracellular adhesion molecule-1 expression in endothelial cells through the nuclear factor-κB pathway, but not through the p38 MAPK pathway. Journal of Periodontal Research 46: 31–38.
Xu, W., Y. Pan, Q. Xu, Y. Wu, J. Pan, J. Hou, L. Lin, X. Tang, C. Li, J. Liu, and D. Zhang. 2018. Porphyromonas gingivalis ATCC 33277 promotes intercellular adhesion molecule-1 expression in endothelial cells and monocyte-endothelial cell adhesion through macrophage migration inhibitory factor. BMC Microbiology 18: 16.
Lue, H., M. Dewor, L. Leng, R. Bucala, and J. Bernhagen. 2011. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cellular Signalling 23: 135–144.
Tillmann, S., J. Bernhagen, and H. Noels. 2013. Arrest Functions of the MIF Ligand/Receptor Axes in Atherogenesis. Frontiers in Immunology 4: 115.
Jayaprakash, K., I. Demirel, S. Gunaltay, H. Khalaf, and T. Bengtsson. 2017. PKC, ERK/p38 MAP kinases and NF-kappaB targeted signalling play a role in the expression and release of IL-1β and CXCL8 in Porphyromonas gingivalis-infected THP1 cells.APMIS 125: 623–633.
Klasen, C., K. Ohl, M. Sternkopf, I. Shachar, C. Schmitz, N. Heussen, E. Hobeika, E. Levit-Zerdoun, K. Tenbrock, M. Reth, J. Bernhagen, and O. El Bounkari. 2014. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. Journal of Immunology 192: 5273–5284.
Schwartz, V., A. Kruttgen, J. Weis, C. Weber, T. Ostendorf, H. Lue, and J. Bernhagen. 2012. Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. European Journal of Cell Biology 91: 435–449.
Lin, S.G., X.Y. Yu, Y.X. Chen, X.R. Huang, C. Metz, R. Bucala, C.P. Lau, and H.Y. Lan. 2000. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circulation Research 87: 1202–1208.
Müller, I., T. Schönberger, M. Schneider, O. Borst, M. Ziegler, P. Seizer, C. Leder, K. Müller, M. Lang, F. Appenzeller, O. Lunov, B. Büchele, M. Fahrleitner, M. Olbrich, H. Langer, T. Geisler, F. Lang, M. Chatterjee, J.F. de Boer, U.J. Tietge, J. Bernhagen, T. Simmet, and M. Gawaz. 2013. Gremlin-1 is an Inhibitor of Macrophage Migration Inhibitory Factor and Attenuates Atherosclerotic Plaque Growth in ApoE (-/-) Mice. Journal of Biological Chemistry 288: 31635–31645.
Bernhagen, J., R. Krohn, H. Lue, J.L. Gregory, A. Zernecke, R.R. Koenen, M. Dewor, I. Georgiev, A. Schober, L. Leng, T. Kooistra, G. Fingerle-Rowson, P. Ghezzi, R. Kleemann, S.R. McColl, R. Bucala, M.J. Hickey, and C. Weber. 2007. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature Medicine 13: 587–596.
Schmitz, C., H. Noels, O. El Bounkari, E. Straussfeld, R.T.A. Megens, M. Sternkopf, S. Alampour-Rajabi, C. Krammer, P.V. Tilstam, N. Gerdes, C. Bürger, A. Kapurniotu, R. Bucala, J. Jankowski, C. Weber, and J. Bernhagen. 2018. Mif-deficiency favors an atheroprotective autoantibody phenotype in atherosclerosis. FASEB Journal 32: 4428–4443.
Rath, S.K., M. Mukherjee, R. Kaushik, S. Sen, and M. Kumar. 2014. Periodontal pathogens in atheromatous plaque. Indian Journal of Pathology and Microbiology 57: 259–264.
Olsen, I., and A. Progulske-Fox. 2015. Invasion of Porphyromonas gingivalis strains into vascular cells and tissue. Journal of Oral Microbiology 7: 28788.
Poole, S., S.K. Singhrao, S. Chukkapalli, M. Rivera, I. Velsko, L. Kesavalu, and S. Crean. 2015. Active Invasion of Porphyromonas gingivalis and Infection-Induced Complement Activation in ApoE (-/-) Mice Brains. Journal of Alzheimers Disease 43: 67–80.
Zernecke, A., J. Bernhagen, and C. Weber. 2008. Macrophage migration inhibitory factor in cardiovascular disease. Circulation 117: 1594–1602.
Leng, L., C.N. Metz, Y. Fang, J. Xu, S. Donnelly, J. Baugh, T. Delohery, Y. Chen, R.A. Mitchell, and R. Bucala. 2003. MIF signal transduction initiated by binding to CD74. Journal of Experimental Medicine 197: 1467–1476.
Su, H., N. Na, X. Zhang, and Y. Zhao. 2017. The biological function and significance of CD74 in immune diseases. Inflammation Research 66: 209–216.
Wan, W., and P.M. Murphy. 2013. Regulation of Atherogenesis by Chemokines and Chemokine Receptors. Archivum Immunologiae et Therapiae Experimentalis (Warsz) 61: 1–14.
Charo, I.F., and R.M. Ransohoff. 2006. The many roles of chemokines and chemokine receptors in inflammation. New England Journal of Medicine 354: 610–621.
Simons, D., G. Grieb, M. Hristov, N. Pallua, C. Weber, J. Bernhagen, and G. Steffens. 2011. Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. Journal of Cellular and Molecular Medicine 15: 668–678.
Schwartz, V., H. Lue, S. Kraemer, J. Korbiel, R. Krohn, K. Ohl, R. Bucala, C. Weber, and J. Bernhagene. 2009. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Letters 583: 2749–2757.
Funding
This study was supported by grants from National Natural Science Foundation of China (nos. 81400518, 81500869). The funders had no role in the study design, data collection, and analysis or in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conception and design of study: ZD, PY. Cell culture, Western blot, qRT-PCR, monocyte-endothelial cell adhesion assay, immunofluorescence staining, and coimmunoprecipitation assays: WY, XW, HJ. P. gingivalis culture and Western blot analysis: WY, XW, LY, LR. Acquisition of data: LJ, LC. Analysis and interpretation: PY, ZD, TX, LL. Writing the manuscript: WY, ZD, PY. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for Publication
Not applicable.
Ethics Approval and Consent to Participate
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wu, Y., Xu, W., Hou, J. et al. Porphyromonas gingivalis-Induced MIF Regulates Intercellular Adhesion Molecule-1 Expression in EA.hy926 Cells and Monocyte-Endothelial Cell Adhesion Through the Receptors CD74 and CXCR4. Inflammation 42, 874–883 (2019). https://doi.org/10.1007/s10753-018-0942-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-018-0942-0