
Abstract
Astrocytes play an important role in immune regulation in the central nervous system (CNS). Dexmedetomidine (DEX) has been reported to exert anti-inflammatory effects on astrocytes stimulated by lipopolysaccharide (LPS) both in vitro and in vivo studies. However, the underlying molecular mechanisms remain poorly understood. This study was designed to evaluate the effects of DEX on tumor necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6) gene expressions in LPS-challenged astrocytes. Moreover, c-Jun N-terminal kinases (JNKs) and p38 mitogen-activated protein kinase (MAPK) pathways in LPS-challenged astrocytes were also investigated. In the present study, astrocytes were stimulated with LPS in the absence and presence of various concentrations of DEX. With real-time PCR assay, we found that LPS significantly increased expressions of TNF-α and IL-6 in mRNA level; however, these effects could be attenuated by DEX. Furthermore, JNK pathway might be involved in LPS-induced astrocyte activation because JNK phosphorylation was significantly increased, and the inhibition of this pathway mediated by DEX as well as SP600125 (JNK inhibitor) decreased TNF-α and IL-6 expressions. Moreover, p38 MAPK was also activated by LPS; however, this pathway seemed to have not participated in DEX-mediated LPS-induced inflammation. These results, taken together, suggest that JNK rather than p38 MAPK signal pathway, provides the potential target for the therapeutic effects of DEX for neuronal inflammatory reactions.
Similar content being viewed by others


INTRODUCTION
Astrocytes are the most abundant cells in the mammalian central nervous system (CNS). Under normal conditions, astrocytes maintain homeostasis in the CNS to support the survival and function of neurons. However, once activated, astrocytes react promptly to the injury, leading to activation of astroglia or astrogliosis [1] and release a diverse set of proinflammatory factors, such as TNF-α, IL-6, and IL-1β [2–4]. Sustained glial inflammatory responses might contribute to the pathophysiology of not only Parkinson’s disease but also other neuronal diseases [5].
Lipopolysaccharide (LPS), a component of gram-negative bacterial cell walls, is one of the most potent activators for regulating gene expression of inflammatory cytokines in experimental animals and humans [6–8]. Previous studies have reported that LPS could stimulate astrocytes and macrophages, inducing excessive production of NO, TNF-α, COX-2, and IL-1β [9, 10].
Dexmedetomidine (DEX), a highly selective and potent α2-adrenoreceptor agonist, provides excellent sedation and analgesia with minimal cardiovascular effects [11]. Numerous studies have shown that DEX is an effective baseline sedative with less opioid requirement and respiratory depression [12, 13]. Available data also suggest that DEX exerts anti-inflammatory effects in animal models of ischemia-reperfusion and ventilator-induced lung injury [14–17].
Besides sedative and analgesic effects, low concentrations of DEX have consistently been found to exert neuroprotective effects in experimental cerebral ischemia and excitotoxic neuronal injury [18, 19]. The target cells in the CNS displaying postjunctional α2-adrenoceptors include astrocytes [20], which mainly express the α2A/D-adrenoceptor subtype [21]. A growing body of evidence demonstrates that astrocyte cells play a critical role in neuronal protection during ischemic injury, ammonia toxicity, and stroke [22–26]. Additionally, DEX significantly increased astrocytes’ release of glial cell line-derived neurotrophic factor (GDNF), inducing subsequent neuroprotective effects in vitro and in vivo [27, 28]. However, the detailed mechanisms for the neuroprotective effects of DEX in astrocytes have not been elucidated. Thus, the primary aim of the current study was to assess the effects of DEX on LPS-induced astrocyte activation and elucidate its possible mechanisms linked to its neuroprotective properties.
MATERIALS AND METHODS
Culture of Primary Astrocyte Cells
Primary rat astrocyte cultures were prepared as previously described [9] with a slight modification from postnatal day 1 to day 2 using Sprague–Dawley (SD) rats. Briefly, the fetal rats were decapitated and hippocampal tissues were collected. The tissues were harvested and digested using 0.25 % trypsin in Hank’s balanced salt solution (HBSS) at 37 °C for 30 min. Trypsin was neutralized with 10 % fetal bovine serum (FBS), and the cell suspension was washed three times in HBSS, decanted, and sequentially passed through a 100 and 40-μm meth and resuspended in Dulbecco’s modified Eagle’s medium (DMEM). The medium was changed every 4 to 5 days with the incubation medium supplemented with 10 % FBS. After 10 days of incubation, flasks were gently shaken for 2 h to loosen weakly attached cells. The supernatant was discarded, and the remaining astrocyte adherent monolayer was detached with 0.25 % trypsin and seeded at 5 × 105 cells/well in six-well plates with incubation medium. Cells were further cultured for 3 days before the experiment. The purity of astrocyte cells was confirmed to be >95 % using immunostaining for glial fibrillary acidic protein (GFAP) antibody. Before experimentation, the medium was replaced with serum-free DMEM containing either PBS or experimental agents. For PCR and Western blot analysis experiments, cells were plated in six-well plates at 5 × 105 cells/well.
Cell Viability Assay
Astrocytes were seeded in a 96-well plate at 2 × 104 cells/well and then kept in a 5 % CO2 incubator at 37 °C overnight. The next day, serial dilutions of DEX and LPS (dissolved in DMEM) were added to the cells to obtain the required final concentration and then incubated for 24 h. The cells were then washed once with PBS and a solution (5 mg/ml) of MTT was added to each well. After 4-h incubation, the MTT solution was discarded carefully and 100-μl dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals. The amount of formazan of each well was determined spectrophotometrically by measuring the absorbance at 570 nm in a plate reader, and each concentration was tested in triplicate. The percentage of cell viability was calculated as the absorbance of treated cells/controls × 100 %.
Real-Time Polymerase Chain Reaction
Total RNA was extracted from primary astrocyte cells by using RNAiso Plus (TaKaRa Bio, Tokyo, Japan) and converted to cDNA by PrimeScript RT reagent kit with gDNA Eraser (TaKaRa Bio). A 2-μg RNA was reverse transcribed and was PCR-amplified using the Access RT-PCR system (Promega, USA) according to the manufacturer’s instructions. Real-time PCR using Takara (Takara, Japan) was carried out on the 7300 System (ABI) for the detection of PCR products.
PCR primers were as follows: TNF-α, forward 5′-CCAGACCCTCACACTCAGATCA-3′ and reverse 5′-GGAGGCTGACTTTCTCCTGGTA-3′; IL-6, forward 5′-CCACCTCACAAGTCGGAGGCTTA-3′ and reverse 5′-GTGCATCATCGCTGTTCATACAATC-3′; and β-actin, forward 5′-TTGTAACCAACTGGGACGATATGG-3′ and reverse 5′-GATCTTGATCTTCATGGTGCTAG-3′. PCR reaction was applied: denaturation at 95 °C for 30 s, 40 cycles of denaturation at 95 °C for 5 s and extension at 60 °C for 31 s, then denaturation at 95 °C for 15 s and extension at 60 °C for 60 s, and denaturation at 95 °C for 15 s. β-Actin was used as a housekeeping gene. TNF-α and IL-6 mRNA expressions were normalized to corresponding β-actin amplicon and quantified using the comparative CT (ΔΔCT) method.
Western Blot Analysis
Cellular proteins were extracted from the primary astrocyte cells using RIPA buffer (Sigma). The protein concentration in the supernatant fluid of the lysate was measured by BCA protein assay (Pierce, Rockfold, IL). Equal amounts of protein in each well were loaded for electrophoresis in 12 % sodium dodecyl sulfate-polyacrylamide gels, and then the gels were transferred to polyvinylidene fluoride (PVDF) microporous membranes (Millipore, Bedford, MA). Membranes containing the transferred proteins were blocked with 5 % skim milk in TBS containing 0.1 % Tween-20 (TBST) for 2 h at room temperature and then incubated overnight with rabbit anti-phospho-c-Jun N-terminal kinase (JNK) antibody (1:500; Cell Signaling, Beverly, MA), anti-JNK antibody (1:500; Cell Signaling), anti-phospho-p38 antibody (1:500; Cell Signaling), anti-p38 antibody (1:500; Cell Signaling), and anti-β-actin antibody (1:2,000; Sigma). Immunoreactivity was detected using horseradish peroxidase conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch, West Grove, PA), visualized by enhanced chemiluminescence detection film. Band density was determined using image analysis system (Image-Pro Plus version 6.0, Media Cybernetics, Silver Spring, MD, USA).
Statistical Analysis
All the experiments were performed at least three times independently. Results were represented as mean ± SEM and performed in triplicate. The data were analyzed using ANOVA with Graphpad Prism 5 software, and Newman–Keuls multiple comparison test was used for post hoc analysis. A value of P < 0.05 was considered statistically significant.
RESULTS
Effects of DEX and LPS on Cell Viability in Astrocytes
MTT assay was used to evaluate the toxic effects of DEX and/or LPS on astrocytes. Astrocytes were incubated with DEX (0.01, 0.1, 1, and 10 μM) and/or LPS (1 μg/ml) for 24 h, and then cell viability was detected by MTT assay. Our results indicated that DEX and 1-μg/ml LPS exert no obvious toxic effects on astrocytes (Fig. 1).

The effects of DEX and LPS on cell viability in cultured primary astrocytes. Astrocyte cells were exposed to different concentrations of DEX (0.01, 0.1, 1, and 10 μM) and/or LPS (1 μg/ml) for 24 h. Cell viability was determined using a colorimetric method. Each data point represents mean ± SEM of at least three separate experiments in which treatments were performed in quadruplicates.
DEX Regulates the Expressions of TNF-α and IL-6 in LPS-Stimulated Astrocytes
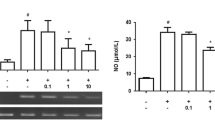
According to our preliminary test, activation of astrocytes by LPS at 1 μg/ml induced a significant and marked increase in TNF-α and IL-6 expressions (data not shown). Therefore, this dose was employed in the present experiment. TNF-α and IL-6 were subjected to real-time PCR. As depicted in Fig. 2a, b, the expressions of TNF-α and IL-6 in primary astrocytes were markedly increased when compared with the control group. The cells were preincubated with various concentrations of DEX for 30 min, followed by the addition of LPS. Our results showed that DEX significantly inhibited TNF-α and IL-6 expressions in a dose-dependent manner, and DEX below 0.1 μM did not show inhibitory effects (Fig. 2c, d).

DEX attenuates TNF-α and IL-6 gene expressions in LPS-activated primary astrocytes. a, b Primary astrocytes were treated with LPS (1 μg/ml) for 1, 2, 6, 12, and 24 h, then TNF-α and IL-6 mRNA expressions were detected by real-time PCR. The expression levels of TNF-α and IL-6 in astrocytes reached the peak at approximately 6 h after LPS stimulation. c, d Primary astrocytes were treated with DEX (0.01, 0.1, 1, and 10 μM) or PBS for 30 min, followed by LPS (1 μg/ml) for 6 h. DEX attenuated LPS-induced TNF-α and IL-6 mRNA expressions at a dose of 0.1 μM or higher, while 0.01-μM DEX showed no inhibitory effect. TNF-α and IL-6 mRNA expressions were normalized to corresponding β-actin amplicon and quantified using the ΔΔCT method. Data represent mean ± SEM of the mean of three independent experiments (**P < 0.01 compared with the corresponding control group; # P < 0.05; ## P < 0.01 compared with the LPS group; n = 3).
DEX Regulates LPS-Induced Inflammatory Responses by the Inhibition of JNK Activation
As JNK and p38 mitogen-activated protein kinase (MAPK) pathways were closely associated with inflammation, we speculated whether DEX exerts its effects on these signals. Our results suggested that LPS significantly increased JNK and p38 phosphorylation (Fig. 3), and the phospho-JNK and phospho-p38 levels were higher after 1 h and then gradually returned to normal (Fig. 3c, d). The total level of JNK and p38 MAPK expressions showed no marked alteration (Fig. 3a, b). Therefore, the time point of 1 h was selected to observe the effect of DEX on phospho-JNK and phospho-p38.

LPS increases JNK and p38 expressions in astrocytes. Primary astrocytes were treated with 1-μg/ml LPS from 30 min to 24 h. Cell lysates were analyzed by Western blotting with antibodies to phospho-JNK, total JNK, phospho-p38 MAPK, total p38 MAPK, and β-actin. Astrocytes treated with LPS showed a marked increase in both phospho-JNK and phospho-p38 MAPK levels. Levels of total JNK and total p38 MAPK remained unchanged. Western blot data were quantified and analyzed as bands of phospho-JNK and phospho-p38 MAPK comparing LPS treatment. LPS significantly increased phospho-JNK and phospho-p38 MAPK, peaked at 1 h, and returned slowly (**P < 0.01 compared with control group, each data represents mean ± SEM at least three separate experiments).
Astrocytes were pretreated with DEX for 30 min, followed by the addition of LPS. It indicated that DEX substantially inhibited JNK activation induced by LPS (Fig. 4a, c). However, DEX increased p38 MAPK phosphorylation (Fig. 4b, d).

DEX attenuates LPS induced inflammatory responses. Primary astrocytes were treated with various concentrations of DEX (0.01, 0.1, 1, and 10 μM) for 30 min followed by stimulation of LPS for 1 h. Cells treated with DEX (0.1, 1, and 10 μM) and LPS showed reduced phospho-JNK activation compared with LPS alone. Total levels of JNK, p38, and phospho-p38 MAPK protein were unchanged. Western blot data were quantified and analyzed as bands of phospho-JNK and phospho-p38 (mean ± SEM; **P < 0.01 compared with the corresponding control group; ## P < 0.01 compared with LPS group; n = 3).
To assess the relationships between JNK and inflammation, SP600125, the JNK antagonist, was used. As astrocytes treated both with LPS and DEX expressed less TNF-α than LPS only, we explored whether DEX exerted its function on JNK signal pathway and subsequently resulted in inhibition of inflammatory factor secretion. Our laboratory has confirmed that astrocytes treated with LPS and SP600125 accumulated significantly less TNF-α and IL-6 in a dose-dependent manner when compared to cultures treated with LPS alone (data not shown). Cells stimulated with 10-μM SP600125 accumulated 34 % less TNF-α than cells treated with LPS alone; cells stimulated with 0.1-μM DEX alone accumulated 26 % less TNF-α than cells treated with LPS alone. Cells treated with 10-μM SP600125 and 0.1-μM DEX alongside LPS stimulation accumulated 49 % less TNF-α than cells treated with LPS alone and significantly less than cells treated with either drug alone. Similar results were observed in IL-6 gene expression (Fig. 5b). Therefore, RT-PCR results indicated that a combinational treatment of SP600125 and DEX can additively suppress LPS-induced TNF-α and IL-6 expressions (Fig. 5).

JNK modulates inflammatory responses in primary astrocyte cells. Primary astrocytes were treated with 0.1-μM DEX and/or SP600125 (JNK inhibitor). Each alone showed attenuation of LPS-induced TNF-α and IL-6 gene expressions; in combination, they showed significant additive effects. Expressions of TNF-α and IL-6 were measured by real-time PCR. Combined DEX with SP600125 reduced LPS-induced TNF-α and IL-6 expressions (**P < 0.01 compared with the corresponding control group; # P < 0.05; ## P < 0.01 compared with the indicated group; n = 3).
DISCUSSION
Our current study demonstrates that DEX exerts anti-inflammatory effects in primary astrocytes and JNK signaling pathway might be involved in DEX-regulated anti-inflammatory properties because high concentrations of DEX (0.1, 1, and 10 μM) attenuated LPS-induced inflammatory responses by modulating JNK activation in primary astrocytes. However, a low concentration of DEX (0.01 μM) caused no significant changes in TNF-α and IL-6 expressions.
In our study, 1-μg/ml LPS was used to stimulate primary astrocytes, and TNF-α as well as IL-6 expressions peaked at 6 h after treatment. These findings are inconsistent with a previous study demonstrating that LPS-induced astrocyte inflammatory responses peaked at 12 h [29]. The divergent results obtained may be related to the differential dose of LPS application, 0.01-μg/ml LPS was employed in their study, and a higher dose of LPS contributed to the activation of astrocytes in a shorter time.
DEX is widely used in ICU sedation, and its pharmacokinetics appears to be highly variable during intensive care [30]. DEX can cross the blood brain barrier and exert many functions on the CNS [31, 32]. The clinically relevant plasma concentration of DEX is 2–20 nM, and the use of DEX at high concentration (0.1 μM) may be clinically practicable [17, 33, 34]. DEX can modulate inflammatory responses and exhibit anti-apoptotic properties via a direct effect on the α2-adrenergic receptors in vitro and in vivo [27, 35, 36]. Several lines of studies have indicated that DEX improves neuronal survival after transient global or focal cerebral ischemia in rats [37, 38]. DEX effectively decreased neuronal damage in a gerbil model of global cerebral ischemia [39] and in a rabbit focal model of ischemia [40]. In animal model of stoke, through α2A-adrenergic receptors, DEX may activate astrocytes and promote GDNF release to protect neurons [26, 41]. Moreover, DEX suppressed spinal glial activation in a rat model of monoarthritis through α2A-adrenergic receptors [42].
Our results showed that DEX (0.1 μM or higher) exhibited potent activity in inhibiting both TNF-α and IL-6 in LPS-stimulated primary astrocytes, though low concentration (0.01 μM) of DEX did not affect TNF-α and IL-6 expressions. The observations suggest that DEX attenuates the excessive inflammatory responses of LPS-induced astrocytes, and these results are in accordance with studies indicating that DEX is a potent suppressor of CNS inflammation [17, 34].
MAPKs are a family of evolutionarily conserved proteins that play critical roles in transducing extracellular stimuli into intracellular responses [28]. This family consists of three major classes: the extracellular signal-regulated kinases (ERKs), the JNKs, and the p38 kinases. ERK is involved in adhesion, proliferation, and cell progression [43], JNK and p38 are involved in apoptosis [44]. Several studies have shown that JNK but not p38 or ERK1/2 was required for the LPS-induced expression of TNF-α mRNA [45, 46]. It was also reported that JNK is involved in LPS-induced NO production [8]. In the present work, it was suggested that JNK but not p38 is involved in the anti-inflammatory properties mediated by DEX. Additionally, other studies have suggested that SP600125 at 5–20 μM blocked part of IL-1β, IL-6, and COX-2 protein expressions [5, 6]. Thus, 10-μM SP600125 was chosen as the administered dosage to evaluate the role of JNK signaling pathway in DEX’s anti-inflammatory properties.
Our results demonstrate that LPS activated JNK and p38 MAPK pathways, inducing inflammatory responses. This process could be that LPS binded toll-like receptor 4(TLR-4) on the cell surface [47], activating phospho-JNK, leading to TNF-α and IL-6 mRNA synthesis. TLR4 was traditionally recognized as the primary receptor for LPS; upon activation, it was involved in both TNF-α and IL-6 releases induced by LPS in astrocytes [48].
DEX significantly inhibited JNK as well as LPS-induced inflammatory responses in primary astrocytes. As a highly selective α2-adrenoreceptor agonist, DEX binds α2-adrenoreceptors on the cell surface and signals a decrease in JNK activation. The decrease in JNK activation of DEX suppresses TNF-α and IL-6 synthesis and exerts anti-inflammatory effects. Our findings also indicate that DEX in combination with specific JNK inhibitor (SP600125) enhanced attenuation of LPS-induced TNF-α and IL-6 expressions in primary astrocytes, suggesting that JNK signaling cascade is a crucial mediator for LPS-induced TNF-α and IL-6 production.
In conclusion, our data show that the clinically relevant concentration of DEX attenuates TNF-α and IL-6 gene expressions in LPS-activated astrocytes and JNK pathway might play an important role in inflammatory or anti-inflammatory effects mediated by LPS or DEX, providing the potential target for the therapeutic effects of DEX for neuronal inflammatory reactions.
REFERENCES
Eng, L.F., and R.S. Ghirnikar. 1994. GFAP and astrogliosis. Brain Pathology 4: 229–237.
Aschner, M. 1998. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology 19: 269–281.
Aschner, M., U. Sonnewald, and K.H. Tan. 2002. Astrocyte modulation of neurotoxic injury. Brain Pathology 12: 475–481.
Munoz-Fernandez, M.A., and M. Fresno. 1998. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Progress in Neurobiology 56: 307–340.
Barcia, C., C.M. Ros, V. Annese, A. Gomez, F. Ros-Bernal, D. Aguado-Llera, M.E. Martinez-Pagan, V. de Pablos, E. Fernandez-Villalba, and M.T. Herrero. 2012. IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Disease 3: e379.
Sato, S., O. Takeuchi, T. Fujita, H. Tomizawa, K. Takeda, and S. Akira. 2002. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. International Immunology 14: 783–791.
Choi, Y., M.K. Lee, S.Y. Lim, S.H. Sung, and Y.C. Kim. 2009. Inhibition of inducible NO synthase, cyclooxygenase-2 and interleukin-1beta by torilin is mediated by mitogen-activated protein kinases in microglial BV2 cells. British Journal of Pharmacology 156: 933–940.
Pocivavsek, A., M.P. Burns, and G.W. Rebeck. 2009. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 57: 444–453.
Gorina, R., T. Santalucia, V. Petegnief, A. Ejarque-Ortiz, J. Saura, and A.M. Planas. 2009. Astrocytes are very sensitive to develop innate immune responses to lipid-carried short interfering RNA. Glia 57: 93–107.
Zhang, X., J. Feng, P. Zhu, and Z. Zhao. 2013. Ketamine inhibits calcium elevation and hydroxyl radical and nitric oxide production in lipopolysaccharide-stimulated NR8383 alveolar macrophages. Inflammation 36: 1094–1100.
Bhana, N., K.L. Goa, and K.J. McClellan. 2000. Dexmedetomidine. Drugs 59: 263–268. discussion 269-270.
Candiotti, K.A., S.D. Bergese, P.M. Bokesch, M.A. Feldman, W. Wisemandle, and A.Y. Bekker. 2010. Monitored anesthesia care with dexmedetomidine: a prospective, randomized, double-blind, multicenter trial. Anesthesia and Analgesia 110: 47–56.
Cooper, L., K. Candiotti, C. Gallagher, E. Grenier, K.L. Arheart, and M.E. Barron. 2011. A randomized, controlled trial on dexmedetomidine for providing adequate sedation and hemodynamic control for awake, diagnostic transesophageal echocardiography. Journal of Cardiothoracic and Vascular Anesthesia 25: 233–237.
Gu, J., J. Chen, P. Xia, G. Tao, H. Zhao, and D. Ma. 2011. Dexmedetomidine attenuates remote lung injury induced by renal ischemia-reperfusion in mice. Acta Anaesthesiologica Scandinavica 55: 1272–1278.
Polat, B., Y. Albayrak, B. Suleyman, H. Dursun, F. Odabasoglu, M. Yigiter, Z. Halici, and H. Suleyman. 2011. Antiulcerative effect of dexmedetomidine on indomethacin-induced gastric ulcer in rats. Pharmacological Reports 63: 518–526.
Yang, C.L., C.H. Chen, P.S. Tsai, T.Y. Wang, and C.J. Huang. 2011. Protective effects of dexmedetomidine-ketamine combination against ventilator-induced lung injury in endotoxemia rats. The Journal of Surgical Research 167: e273–e281.
Peng, M., Y.L. Wang, C.Y. Wang, and C. Chen. 2012. Dexmedetomidine attenuates lipopolysaccharide-induced proinflammatory response in primary microglia. The Journal of Surgical Research 179: e219–e225.
Bell, M.T., F. Puskas, D.T. Bennett, P.S. Herson, N. Quillinan, D.A. Fullerton, and T.B. Reece. 2013. Dexmedetomidine, an alpha-2a adrenergic agonist, promotes ischemic tolerance in a murine model of spinal cord ischemia-reperfusion. Journal of Thoracic and Cardiovascular Surgery. doi:10.1016/j.jtcvs.2013.07.043.
Sato, K., T. Kimura, T. Nishikawa, Y. Tobe, and Y. Masaki. 2010. Neuroprotective effects of a combination of dexmedetomidine and hypothermia after incomplete cerebral ischemia in rats. Acta Anaesthesiologica Scandinavica 54: 377–382.
Ebersolt, C., M. Perez, and J. Bockaert. 1981. Alpha 1 and alpha 2 Adrenergic receptors in mouse brain astrocytes from primary cultures. Journal of Neuroscience Research 6: 643–652.
Enkvist, M.O., H. Hamalainen, C.C. Jansson, J.P. Kukkonen, R. Hautala, M.J. Courtney, and K.E. Akerman. 1996. Coupling of astroglial alpha 2-adrenoreceptors to second messenger pathways. Journal of Neurochemistry 66: 2394–2401.
Panickar, K.S., and M.D. Norenberg. 2005. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia 50: 287–298.
Rao, K.V., K.S. Panickar, A.R. Jayakumar, and M.D. Norenberg. 2005. Astrocytes protect neurons from ammonia toxicity. Neurochemical Research 30: 1311–1318.
Giaume, C., A. Koulakoff, L. Roux, D. Holcman, and N. Rouach. 2010. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nature Review Neuroscience 11: 87–99.
Giaume, C. 2010. Astroglial wiring is adding complexity to neuroglial networking. Frontiers in Neuroenergetics. doi:10.3389/fnene.2010.00129.
Gleichman, A.J., and S.T. Carmichael. 2013. Astrocytic therapies for neuronal repair in stroke. Neuroscience Letters. doi:10.1016/j.neulet.2013.10.055.
Engelhard, K., C. Werner, E. Eberspacher, M. Bachl, M. Blobner, E. Hildt, P. Hutzler, and E. Kochs. 2003. The effect of the alpha 2-agonist dexmedetomidine and the N-methyl-D-aspartate antagonist S(+)-ketamine on the expression of apoptosis-regulating proteins after incomplete cerebral ischemia and reperfusion in rats. Anesthesia and Analgesia 96: 524–531.
Chen, Z., T.B. Gibson, F. Robinson, L. Silvestro, G. Pearson, B. Xu, A. Wright, C. Vanderbilt, and M.H. Cobb. 2001. MAP kinases. Chemistry Review 101: 2449–2476.
Kozuka, N., Y. Kudo, and M. Morita. 2007. Multiple inhibitory pathways for lipopolysaccharide- and pro-inflammatory cytokine-induced nitric oxide production in cultured astrocytes. Neuroscience 144: 911–919.
Iirola, T., R. Laitio, E. Kentala, R. Aantaa, J.P. Kurvinen, M. Scheinin, and K.T. Olkkola. 2010. Highly variable pharmacokinetics of dexmedetomidine during intensive care: a case report. Journal of Medical Case Reports 4: 73.
Hayashi, Y., K. Sumikawa, M. Maze, A. Yamatodani, T. Kamibayashi, M. Kuro, and I. Yoshiya. 1991. Dexmedetomidine prevents epinephrine-induced arrhythmias through stimulation of central alpha 2 adrenoceptors in halothane-anesthetized dogs. Anesthesiology 75: 113–117.
Benggon, M., H. Chen, R. Applegate, R. Martin, and J.H. Zhang. 2012. Effect of dexmedetomidine on brain edema and neurological outcomes in surgical brain injury in rats. Anesthesia and Analgesia 115: 154–159.
Ebert, T.J., J.E. Hall, J.A. Barney, T.D. Uhrich, and M.D. Colinco. 2000. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology 93: 382–394.
Lai, Y.C., P.S. Tsai, and C.J. Huang. 2009. Effects of dexmedetomidine on regulating endotoxin-induced up-regulation of inflammatory molecules in murine macrophages. The Journal of Surgical Research 154: 212–219.
Engelhard, K., C. Werner, S. Kaspar, O. Mollenberg, M. Blobner, M. Bachl, and E. Kochs. 2002. Effect of the alpha2-agonist dexmedetomidine on cerebral neurotransmitter concentrations during cerebral ischemia in rats. Anesthesiology 96: 450–457.
Taniguchi, T., Y. Kidani, H. Kanakura, Y. Takemoto, and K. Yamamoto. 2004. Effects of dexmedetomidine on mortality rate and inflammatory responses to endotoxin-induced shock in rats. Critical Care Medicine 32: 1322–1326.
Bekker, A., and M.K. Sturaitis. 2005. Dexmedetomidine for neurological surgery. Neurosurgery 57: 1–10.
Zhang, Y., and H.K. Kimelberg. 2005. Neuroprotection by alpha 2-adrenergic agonists in cerebral ischemia. Current Neuropharmacology 3: 317–323.
Kuhmonen, J., J. Pokorny, R. Miettinen, A. Haapalinna, J. Jolkkonen, P. Riekkinen Sr., and J. Sivenius. 1997. Neuroprotective effects of dexmedetomidine in the gerbil hippocampus after transient global ischemia. Anesthesiology 87: 371–377.
Maier, C., G.K. Steinberg, G.H. Sun, G.T. Zhi, and M. Maze. 1993. Neuroprotection by the alpha 2-adrenoreceptor agonist dexmedetomidine in a focal model of cerebral ischemia. Anesthesiology 79: 306–312.
Yan, M., H. Dai, T. Ding, A. Dai, F. Zhang, L. Yu, G. Chen, and Z. Chen. 2011. Effects of dexmedetomidine on the release of glial cell line-derived neurotrophic factor from rat astrocyte cells. Neurochemistry International 58: 549–557.
Xu, B., W.S. Zhang, J.L. Yang, N. Lu, X.M. Deng, H. Xu, and Y.Q. Zhang. 2010. Evidence for suppression of spinal glial activation by dexmedetomidine in a rat model of monoarthritis. Clinical and Experimental Pharmacology & Physiology 37: e158–e166.
Lenormand, P., G. Pages, C. Sardet, G. L’Allemain, S. Meloche, and J. Pouyssegur. 1993. MAP kinases: activation, subcellular localization and role in the control of cell proliferation. Advances in Second Messenger and Phosphoprotein Research 28: 237–244.
Chen, Y.R., W. Wang, A.N. Kong, and T.H. Tan. 1998. Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis induced by anticarcinogenic isothiocyanates. The Journal of Biological Chemistry 273: 1769–1775.
Swantek, J.L., M.H. Cobb, and T.D. Geppert. 1997. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Molecular and Cellular Biology 17: 6274–6282.
Shen, J., I. Sakaida, K. Uchida, S. Terai, and K. Okita. 2005. Leptin enhances TNF-alpha production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sciences 77: 1502–1515.
Chow, J.C., D.W. Young, D.T. Golenbock, W.J. Christ, and F. Gusovsky. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. The Journal of Biological Chemistry 274: 10689–10692.
Lien, E., T.K. Means, H. Heine, A. Yoshimura, S. Kusumoto, K. Fukase, M.J. Fenton, M. Oikawa, N. Qureshi, B. Monks, R.W. Finberg, R.R. Ingalls, and D.T. Golenbock. 2000. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. Journal of Clinical Investigation 105: 497–504.
ACKNOWLEDGMENTS
This work was funded by the National Science Foundation of China (81202230 and 81273115) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding authors
Additional information
X. Zhang and J. Wang contributed equally to this study.
Rights and permissions
About this article
Cite this article
Zhang, X., Wang, J., Qian, W. et al. Dexmedetomidine Inhibits Tumor Necrosis Factor-Alpha and Interleukin 6 in Lipopolysaccharide-Stimulated Astrocytes by Suppression of c-Jun N-Terminal Kinases. Inflammation 37, 942–949 (2014). https://doi.org/10.1007/s10753-014-9814-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-014-9814-4