
Abstract
Decreased systolic function is a central factor in the pathogenesis of heart failure, yet there are no safe medical therapies to improve cardiac function in patients. Currently available inotropes, such as dobutamine and milrinone, increase cardiac contractility at the expense of increased intracellular concentrations of calcium and cAMP, contributing to increased heart rate, hypotension, arrhythmias, and mortality. These adverse effects are inextricably linked to their inotropic mechanism of action. A new class of pharmacologic agents, cardiac myosin activators, directly targets the kinetics of the myosin head. In vitro studies have demonstrated that these agents increase the rate of effective myosin cross-bridge formation, increasing the duration and amount of myocyte contraction, and inhibit non-productive consumption of ATP, potentially improving myocyte energy utilization, with no effect on intracellular calcium or cAMP. Animal models have shown that this novel mechanism increases the systolic ejection time, resulting in improved stroke volume, fractional shortening, and hemodynamics with no effect on myocardial oxygen demand, culminating in significant increases in cardiac efficiency. A first-in-human study in healthy volunteers with the lead cardiac myosin activator, CK-1827452, as well as preliminary results from a study in patients with stable chronic heart failure, have extended these findings to humans, demonstrating significant increases in systolic ejection time, fractional shortening, stroke volume, and cardiac output. These studies suggest that cardiac myosin activators offer the promise of a safe and effective treatment for heart failure. A program of clinical studies are being planned to test whether CK-1827452 will fulfill that promise.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure is the primary reason for admission in greater than 1 million hospitalizations in the United States alone [1] and 65–70% of these patients have at least a mild decrease in systolic dysfunction [2]. The central role of ventricular dysfunction in the pathogenesis of heart failure has been recognized explicitly for over 50 years, and implicitly since Dr. William Withering’s application of the foxglove treatment (digitalis) developed by an elderly woman in Shropshire, England over 200 year ago. While the neurohormonal model has appropriately emerged as a dominant factor in understanding chronic heart failure, cardiac dysfunction remains a primary and necessary instigating event. In the setting of acute heart failure, altered hemodynamics continue to be a primary target for therapies. Testament to the necessity of these agents may be found in the continued use of intravenous agents with inotropic properties that are known to have significant adverse effects, such as dobutamine and milrinone, [3] and the continued active development of both intravenous and oral agents despite scores of failures. This review will discuss the development of a novel class of agents designed to improve cardiac performance, known as cardiac myosin activators, in the context of the mechanism of cardiac contraction and the currently available agents.
Mechanisms to improve cardiac performance
An in-depth explication of the mechanism of cardiac performance is beyond the scope of this review. However, understanding certain elements of myocardial contractility is essential to appreciate how cardiac myosin activators might be useful in the treatment of heart failure. Myocardial contraction is the result of a remarkable transduction of chemical energy into mechanical energy that is regulated on a beat-to-beat basis by inter-related signaling pathways acting on the sarcomere, or the contractile unit of the myocyte [4]. Sarcomeres have a complicated structure with the main force-generating unit consisting of actin, myosin, and multiple regulatory proteins (such as troponin and tropomyosin). The myosin complex consists of two myosin heavy chains and two pairs of myosin light chains. The myosin heavy chain has a globular head that has an ATPase domain (which cleaves ATP to produce energy), as well as an actin-binding site, through which the contractile force is transduced. Cardiac troponin (cTn) and tropomyosin form a complex that regulates the interaction of myosin with actin in a calcium sensitive manner [5]. In the resting state, there are low calcium concentrations in the myocyte, and the troponin–tropomyosin complex blocks cross-bridge formation between actin and myosin. Depolarization of the myocyte triggers calcium release from the sarcoplasmic reticulum, increasing sarcomeric concentrations of calcium, resulting in calcium binding to cTnC and a shift of tropomyosin so that it no longer blocks actin–myosin cross-bridge formation. Subsequently, myosin binds to actin and, from energy released through the hydrolysis of ATP to ADP, produces a force-generating conformational change and shortening of the sarcomere. Calcium is actively transported back into the sarcoplasmic reticulum in a highly energy-intensive process, awaiting release again after the next depolarization event, a cycle that repeats for the lifetime of the organism.
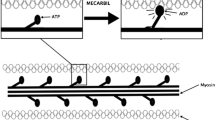
The cardiac actin–myosin cycle [6, 7] is central to this process of myocardial force generation and continued research has elucidated important details of the cycle (see Fig. 1) which are very important to a discussion of cardiac myosin activators. The linking of mechanical and enzymatic cycles of the myosin motor has been the subject of intense investigation for decades culminating in a well-accepted model depicted in Fig. 1. After the binding of ATP to myosin (Fig. 1, Step 1), the myosin head rapidly dissociates from actin. Myosin rapidly hydrolyzes the bound ATP into ADP and Pi (Fig. 1, Step 2), which allows a weak interaction between the myosin head and actin as it scans for a proper binding site (Fig. 1, Step 3). On binding, the actin–myosin–ADP–Pi complex undergoes an activation step (the slowest step in the overall cycle) which simultaneously converts the complex to a strongly bound state and releases the Pi (Fig. 1, Step 4). The release of the Pi results in the transduction step with a conformational “bending” of the myosin head, producing approximately a 10-nm stroke (Fig. 1, Step 5). Subsequently, the release of ADP occurs and the exit from the strongly bound state (Fig. 1, Step 6) is triggered by the rapid binding of ATP to the myosin complex which leads to its dissociation from actin and prepares the cycle to repeat. Two aspects of this cycle are important to note. First, the rate-limiting step in cross-bridge formation and thus, the number of available cross bridges per depolarization (heart beat), is the transition from the weakly bound to the strongly bound state of the actin–myosin complex (Fig. 1, Step 4). Second, ATP is hydrolyzed independent of actual mechanical work (Fig. 1, Step 2) and if there is subsequent actin-independent release of the Pi, no force-generating stroke is produced. A portion of cardiac myosin ATP hydrolysis, or energy consumption, does not result in strong actin–myosin binding and is thus wasted although the rate of actin independent ATP hydrolysis is thought to be slow.

The actin–myosin cycle (Modified from [6])
“Inotrope” versus cardiac myosin activator
Inotropy may be considered an intrinsic or directly evoked improvement of myocardial contractility, as measured by indices of force and the rate of change in variables of ventricular contraction (e.g., volume, pressure, force). The current drugs that target an increase of myocardial contractility share a similar mechanism of action: they increase intracellular cAMP and ultimately calcium (Fig. 2). Catecholamines (isoproterenol, norepinephrine, dobutamine, dopamine) increase cAMP through receptor-mediated increases in cAMP production with multiple downstream signaling effects. Phosphodiesterase (PDE) inhibitors prevent the degradation of cAMP and consequently also increase intracellular concentrations. Both of these mechanisms result in increased intracellular calcium, which in myocytes increases contractility but with significant costs. The increased intracellular calcium needs to be actively transported back into the sarcoplasmic reticulum with each cardiac cycle, markedly increasing the ATP required to maintain calcium homeostasis. This increased energy utilization results in increased myocardial oxygen demand. Myocardial oxygen demand is further exacerbated by an increased heart rate. Elevations in intracellular calcium concentrations and increased calcium transients are also considered to play a role in the increased arrhythmogenicity of these agents. These effects contribute to the observed clinical increase in arrhythmias and mortality with these drugs, especially in the setting of myocardial ischemia [8–11]. All of the currently available agents with inotropic properties also have significant vascular effects, predominantly vasodilation in the case of dobutamine and milrinone. The hypotensive effects of dobutamine and milrinone are particularly dangerous in the very patients in whom these agents are indicated [9, 10]. The challenge for these inodilator agents is that the adverse effects are directly related to and a consequence of their inotropic mechanism of action.

Schematic of myocyte and signaling pathways
As the details of the actin–myosin cycle were elucidated, a potential new approach to increasing cardiac performance emerged. Small molecules that could activate cardiac myosin would theoretically be able to improve the ability of the heart to perform work without altering intracellular cAMP or calcium transients. Ideally, an agent would increase the rate-limiting step in the actin–myosin cycle by increasing the transition from the weakly bound to the strongly bound state (Fig. 1, Step 4), allowing more myosin heads to be active during each heart beat. In addition, if an agent could decrease the actin-independent release of Pi, it would improve myocardial efficiency. A useful analogy for these effects is to picture a multi-oared rowboat or racing shell where some of the crew stroke with their oars out of the water; a cardiac myosin activator would result in more oars in the water and fewer oars stroking out of the water. In the absence of incurring additional energetic costs related to calcium cycling, this mechanism would be the most efficient way of increasing cardiac contractility.
Pre-clinical development of cardiac myosin activators
Starting with a biochemically reconstituted sarcomere to screen in high throughput fashion for small molecule activators of the cardiac sarcomere, a group of biologists led by Dr. Fady Malik, pursued the hypothesis and reported that cardiac myosin activators could improve cardiac performance [12]. The initial hit compounds discovered with this assay were subsequently optimized in an iterative fashion using biochemical assays in conjunction with assessment of contractility in cardiac myocytes. In parallel, the compounds were optimized for drug-like properties (e.g., drug metabolism, pharmacokinetics, solubility), and undesirable activities were ruled out (such as phosphodiesterase III activity and alterations in calcium transients). Ultimately, the best compounds were characterized using in vivo models of cardiac function (rat and dog). Four compounds emerged from this process for which public pre-clinical data are available, one of which has progressed into human clinical development.
Data presented from three pre-clinical compounds supported the hypothesis that cardiac myosin activators could improve cardiac performance independent of changes in the calcium transient. CK-0689705 [13] did not increase calcium transients in isolated rat ventricular myocytes, but did increase fractional myocyte shortening in a dose-dependent manner. Interestingly, despite these increases in contractility, CK-0689705 did not increase the velocity of contraction, in contrast to classic inotropes, such as isoproterenol, which increase contraction velocity and calcium transients. Furthermore, the effect of CK-0689705 on cardiac function was preserved in myocytes from rats with heart failure due to myocardial infarction, in contrast to the decreased responsiveness observed with the beta-adrenergic agonist, isoproterenol.
The second pre-clinical compound, CK-1122534, also supported the hypothesis of cardiac myosin activation. CK-1122534 [14] activated the ATPase of the motor-domain of cardiac myosin (the S1 subunit) in three test systems, using purified cardiac actin and cardiac S1, a calcium-regulated system composed of cardiac S1 and a reconstituted thin filament, and skinned cardiac myofibrils. This myosin activity was confirmed to be cardiac specific, with no effect on skeletal or smooth muscle myosins. Transient kinetic methods were used to demonstrate that CK-1122534 increased by two-fold the rate of actin-dependent Pi release, a measure of the transition from the weakly bound to the strongly bound state (Fig. 1, Step 4) without affecting the rates of other steps in the cycle. In addition, CK-1122534 was shown to overcome the inhibiting effects of BDM (2,3-butanedione-2-monoxime) and blebbistatin on the contractility of cardiac myocytes. Both BDM and blebbistatin [15] inhibit myosin activity by uncoupling myosin–actin cross-bridge contractility from the calcium transient. Thus, at high concentrations of these inhibitors, there is no contractility despite persistent cycling of the calcium transient. CK-1122534 partially restored myocyte contractility in the setting of marked inhibition by both BDM and blebbistatin, also supporting its role as a direct myosin activator. As with CK-0689705, CK-1122534 was also demonstrated to have no phosphodiesterase activity and to not alter calcium transients, while increasing myocyte fractional shortening, even in the presence of beta-adrenergic blockade with propranolol [16].
The third pre-clinical compound, CK-1213296, extended these observations into animal models of heart failure [17]. As with the previously mentioned agents, CK-1213296 was active in multiple experimental systems, increasing the ATPase activity of cardiac myosin specifically without affecting calcium transients. In addition, CK-1213296 was shown to increase actin-dependent Pi release (Fig. 1, Step 4), facilitating the transition from the weak-to-the-strong binding state, as well as to decrease the rate of actin-independent phosphate release, reducing non-productive and hence wasteful hydrolysis of ATP. In anesthetized Sprague-Dawley rats, CK-1213296 increased cardiac fractional shortening by 25% (12% absolute increase), as assessed by echocardiography. In another series of experiments (Fig. 3), mongrel dogs were chronically instrumented with pressure transducers in the left ventricle (LV), left atrium, and aorta, with ultrasound crystals implanted for measurement of LV dimensions and wall thickness, and in some dogs, with an aortic flow probe and inferior vena caval occluder to assess pressure–volume loops. The dogs underwent myocardial infarction using a left anterior descending artery occluder followed by continuous rapid ventricular pacing (220–240 bpm) to produce heart failure. Hemodynamic assessments were made at 5–6 weeks after the infarction procedure and the pacemaker was turned off during these measurements. The dogs demonstrated clear evidence of heart failure with increases in resting heart rate and left atrial pressure, and marked decreases in mean arterial pressure, dP/dt, cardiac output, stroke volume, and fractional shortening. Administration of a bolus followed by an infusion of CK-1213296 resulted in significant improvements in cardiac function (end-systolic elastance, stroke volume, wall thickening, fractional shortening), associated with decreased filling pressures (left atrial pressure) and heart rate. Thus, these experiments with CK-1213296 fulfilled the therapeutic hypothesis that cardiac myosin activators could improve cardiac performance in the setting of heart failure.

Schematic diagram of dog model of heart failure
Development of the cardiac myosin activator, CK-1827452
The previous candidate compounds provided essential experience for the development of the cardiac myosin activator, CK-1827452 (Fig. 4). CK-1827452 increases cardiac myosin ATPase activity in a dose-dependent manner that is selective for cardiac myosin versus non-cardiac myosins and is independent of the sarcomeric regulatory proteins. As seen with previous myosin activators, CK-1827452 accelerates productive phosphate release (actin-dependent Pi release; Fig. 1, Step 4), increasing the transition from the weak to the strong binding state, while inhibiting non-productive phosphate release (actin-independent phosphate release), hence reducing wasted ATP hydrolysis [18, 19]. In cellular preparations, this compound markedly increased myocyte contraction with an associated increase in the time-to-peak contraction, indicating an increased duration of contraction. This finding is a hallmark of these agents where contraction velocity is largely unaltered, but the time spent contracting is increased. Despite these increases in contractility, there are no changes in the calcium transient and there is no alteration of the sarcoplasmic reticulum calcium content or in the sodium calcium exchanger [20]. CK-1827452 was also able to overcome the inhibition of cross-bridge formation by BDM without increasing the calcium transient magnitude and had no evidence of phosphodiesterase activity [21]. All of these studies confirmed that the mechanism of action of CK-1827452 as a specific cardiac myosin activator, but additional studies in animals demonstrated its potential clinical efficacy.

Structure of CK-1827452
The in vivo efficacy of CK-1827452 was studied in two animal species. In anesthetized Sprague-Dawley rats, infusion with the agent acutely increased LV fractional shortening by 20% in a dose-dependent manner with no significant effect on diastolic diameter, as assessed by two-dimensional guided M-mode echocardiography. Another group of rats underwent ligation of the left anterior descending coronary artery and were selected for study on the basis of decreased fractional shortening compared to sham-operated rats. These rats with heart failure had significantly increased heart weights and markedly reduced fractional shortening (from 50% in shams to 22% in heart failure). Administration of CK-1827452 to anesthetized rats with either heart failure or sham operations resulted in a similar dose-related relative increase in fractional shortening (~20%), as compared to baseline. Interestingly, sham-operated animals also demonstrated a significant decrease in diastolic diameter at the highest dose.
Using a similar protocol as described above (Fig. 3), another series of experiments were conducted in the two labs of Drs. Stephen Vatner and David Kass in mongrel dogs with heart failure due to myocardial infarction and continuous rapid ventricular pacing. After 5–6 weeks of post-myocardial infarction pacing to establish heart failure, infusion of CK-1827452 produced significant improvements in multiple parameters at both 15 min and 4 h. There were marked increases in LV wall thickening (30–40% change from baseline), fractional shortening (30–40%), stroke volume (50–60%), and cardiac output (20–25%). These improvements in cardiac function were associated with more favorable hemodynamics, including decreased left atrial pressure, left ventricular end-diastolic pressure, total peripheral resistance, and heart rate with no change in mean arterial or left ventricular systolic pressure. Importantly, there were no significant changes in coronary blood flow, coronary sinus oxygen content, or myocardial oxygen consumption (MVO2). Stroke work increased over 60% in the context of no increase in oxygen demand, resulting in a 25–30% increase in cardiac efficiency. Consistent with the mechanism of action, the systolic ejection time increased, reflecting the increased duration of contraction observed in the pre-clinical studies, with no change in the velocity of contraction (dP/dt). These findings confirmed the therapeutic hypothesis that cardiac myosin activators could improve cardiac function without compromising myocardial oxygen demand given the absence of changes in cellular calcium homeostasis.
Rapid pacing of dogs produces heart failure but without significant left ventricular hypertrophy unlike the situation most common in human heart failure where left ventricular hypertrophy accompanies progressive ventricular dilatation. In addition, since the mechanism of action increases the duration of systole, there were questions of how this drug would perform in the setting of left ventricular hypertrophy and diastolic dysfunction. To address this question, Dr. Stephen Vatner and colleagues performed another series of experiments using a similar instrumentation protocol in mongrel dogs that underwent aortic banding to produce a spectrum of cardiac abnormalities ranging from severe left ventricular hypertrophy to decompensated heart failure (Fig. 5) [22]. In dogs with severe left ventricular hypertrophy without heart failure, CK-1827452 produced an increased systolic ejection time, as well as increased ventricular wall thickening, associated with only transient increases in fractional shortening, and stroke volume. As with prior studies in animals with heart failure, dogs with left ventricular hypertrophy and heart failure had increases in systolic ejection time, systolic wall thickening, fractional shortening and stroke volume in response to CK-1827452, associated with reductions in left ventricular end-diastolic pressure, mean left atrial pressure, and heart rate. Importantly, there was no change in the epicardial-to-endocardial blood flow ratio or myocardial oxygen demand during CK-1827452 infusion as compared to baseline in dogs either with heart failure or left ventricular hypertrophy. Thus, cardiac efficiency again improves in this model of heart failure plus left ventricular hypertrophy. In sum, these animal studies provided the support for the potential benefit of CK-1827452 in the treatment of heart failure and encouraged investigation of this agent in humans.

Schematic diagram of dog model of left ventricular hypertrophy and heart failure
The first-in-human study (CY1111) with CK-1827452 was initially presented as a late-breaking clinical trial at the Heart Failure Society of America by Dr. John Teerlink [23]. The primary objective of the study was to determine the maximally tolerated dose (MTD) and plasma concentrations of CK-1827452 administered as a 6 h intravenous infusion, while secondary objectives included evaluation of its pharmacodynamics (including its effect on left ventricular systolic function as assessed by echocardiography), pharmacokinetics, and safety and tolerability. This study enrolled healthy men in four ascending dose cohorts (n = 34 subjects) who received a 6-h double-blind infusion once each week for four weeks. Each treatment sequence consisted of three ascending CK-1827452 doses and a placebo infusion randomized into each treatment sequence, where the low dose of each subsequent cohort was the high dose from the prior cohort; modifications of the dosing schedule were made as the MTD was achieved (Table 1). CK-1827452 was studied at 0.005, 0.015, 0.025, 0.0625, 0.125, 0.25, 0.5, 0.625, 0.75 and 1.0 mg/kg/h. Transthoracic echocardiograms were obtained at baseline and after 6 h of infusion, as well as other time-points, and over-read without knowledge of time or treatment by a panel of cardiologists. Consistent with its mechanism of action, a linear dose-related increase in LV systolic ejection time was observed, with an almost perfect correlation between plasma concentrations and systolic ejection time (r 2 = 0.97) [24]. At the MTD of 0.5 mg/kg/h, statistically significant increases in LV systolic function were demonstrated with absolute increases in ejection fraction and fractional shortening of 6.8% (P < 0.0001) and 9.2% (P < 0.0001), respectively. These increases in indices of left ventricular function were associated with a mean 84 ms prolongation of systolic ejection time (P < 0.0001), and directly correlated with dose, and even more directly with plasma concentration (r 2 = 0.93 for LV fractional shortening; r 2 = 0.90 for ejection fraction) [24]. Furthermore, there were direct relationships between the systolic ejection time and both measures of ventricular systolic function (r 2 = 0.92 for LV fractional shortening; r 2 = 0.94 for ejection fraction). The very close relationship between the plasma concentrations of CK-1827452 and the systolic ejection time and the improvements in ventricular function suggest that the effect on cardiac function after dosing with this drug may be reliably observed and predictable.
In this first-in-human study, [23, 25] at the maximally tolerated dose (MTD) of 0.5 mg/kg/h for 6 h and below, CK-1827452 was well tolerated when compared to placebo. In addition, CK-1827452 was clinically well tolerated at 0.625 mg/kg/h for 6 h but too few volunteers received this dose to define it according to protocol as the MTD. By design, the study found doses that were not well tolerated; three patients receiving the 0.75 and 1.0 mg/kg/h doses developed signs and symptoms of myocardial ischemia most likely due to excessive prolongation of the systolic ejection time, causing decreased diastolic coronary perfusion and decreased diastolic filling. This anticipated response to the excessive pharmacologic effect of an over-dose resulted in premature discontinuation of study drug infusion in three out of the four patients at these dose levels with rapid and complete resolution of abnormalities. Importantly, no “off-target” adverse effects were noted even at these extreme doses. At doses up to and through 0.5 mg/kg/h, there were no dose-related changes versus placebo in heart rate or the electrocardiographic PR, and the QT or corrected QT intervals actually shorten. Up to and including the MTD, there was no dose-related increase in the overall incidence of adverse events. In this study, the cardiac myosin activator, CK-1827452, increased LV systolic function by a unique mechanism, directly increasing systolic ejection time rather than contraction velocity. These first-in-human data recapitulate the pharmacodynamic effects in normal animals and animals with heart failure, in support of the putative mechanism of action and the potential clinical utility of CK-1827452 in patients with heart failure.
Interim results from the first study of CK-1827452 in patients with heart failure (CY1121) were initially presented by Dr. John Cleland at the European Society of Cardiology Heart Failure Congress [26] and subsequently at the European Society of Cardiology and Heart Failure Society of America [27] meetings. The primary objective of the study was to evaluate the safety and tolerability of CK-1827452 administered as an intravenous infusion to patients with stable heart failure, while secondary objectives included evaluation of its pharmacodynamics (including its effect on left ventricular systolic function as assessed by echocardiography) and pharmacokinetics. Patients were eligible for the study if they had a clinical diagnosis of heart failure, an ejection fraction less than 40%, normal sinus rhythm, and a stable drug regimen, including an ACE inhibitor or ARB, beta blocker, and diuretics, if necessary. Patients with a cardiovascular hospitalization within 6 weeks of entry or CCS class III or IV angina were excluded. Patients were randomized sequentially into each dose cohort, where each patient would receive one of three active ascending CK-1827452 doses with a placebo infusion randomly interspersed in the sequence of doses; doses were separated by at least a week. Loading doses and maintenance doses were modeled on the basis of the pharmacokinetic data derived from the previously described CY1111 study [23]. Importantly, the pharmacokinetics of the healthy volunteers were found to be highly predictive of the pharmacokinetics of the patients with heart failure (see Table 2), and across the range of plasma concentrations evaluated, the pharmacokinetics of CK-1827452 were generally linear with respect to dose.
The CY1121 study is on-going, so the only complete interim data available at this time are from the first three cohorts. As with the healthy volunteer study, there were pronounced, dose- and concentration-dependent increases in the systolic ejection time, supporting the mechanism of action of this cardiac myosin activator, but the proportion of the cardiac cycle dedicated to ejection remained relatively constant, due to the decline in heart rate at higher concentrations. There were concentration-dependent increases in multiple measures of cardiac systolic function (Doppler-derived stroke volume, fractional shortening, and ejection fraction; all P < 0.0001 for correlation with CK-1827452 concentration, except ejection fraction, P < 0.05). The Doppler-derived stroke volume significantly increased by approximately 20% in the setting of a 10% decrease in heart rate, resulting in a marked increase in cardiac output, initially evident at low-to-moderate concentrations. The fractional shortening increased by about 23% at the higher concentrations, while there were no significant increases in ejection fraction although a concentration-dependent trend toward increase did reach statistical significance. In addition, there were statistically significant correlations between CK-1827452 concentration and decreases in left ventricular end-systolic volume (P < 0.05), while decreases in left ventricular end-diastolic volume were not statistically significant. The safety data from this preliminary analysis suggest that CK-1827452 was well tolerated with no serious adverse events reported in heart failure patients exposed to the intended range of doses and plasma concentrations. The interim findings from this first study of CK-1827452 in patients with heart failure clearly demonstrated improvements in cardiac function, which were directly related to the plasma concentration of the drug.
Future directions
The findings from these Phase I and IIa studies provide the basis of considerable enthusiasm for this new pharmacologic class of cardiac myosin activators. Cytokinetics, Inc. has conducted five Phase 1 clinical studies of CK-1827452 in healthy subjects, including a study of oral bioavailability [28]. In this study, each healthy male volunteer (n = 8) received four doses of CK-1827452 (0.125 mg/kg/h intravenous, 0.125 mg/kg oral liquid formulation, 0.125 mg/kg oral capsule fasted, and 0.125 mg/kg oral capsule fed), and blood samples were obtained for thorough pharmacokinetic study. There was no evidence of first-pass metabolism, with absolute bioavailability of all formulations approaching 100% and no effect of food on overall bioavailability. In addition, it was well tolerated in all formulations with no significant safety issues. Its absorption was very rapid though, which led to efforts to develop a modified release formulation. Subsequent studies performed with modified-release prototype formulations led to the selection of one prototype for further development and support the potential of CK-1827452 as an oral therapy for chronic heart failure. Other Phase IIa studies are also underway including a study of the safety and tolerability of intravenous and oral formulations in patients with ischemic cardiomyopathy, and another study in stable heart failure patients undergoing cardiac catheterization to evaluate ventricular performance and myocardial oxygen consumption [29].
Cardiac myosin activators represent a novel therapeutic approach to improving cardiac performance. By accelerating the transition of the actin–myosin complex from the weakly-to-the-strongly bound state and inhibiting the non-productive hydrolysis of ATP, these agents increase the ability of the heart to perform the work of pumping blood without altering calcium homeostasis or increasing cAMP. The absence of these effects on myocyte calcium suggests that cardiac myosin activators may not increase arrhythmias, myocardial oxygen demand, and ischemia, as observed with the currently available inotropes (e.g., milrinone and dobutamine). Studies in rats, dogs, and humans have all supported the hypothesis of this beneficial effect on cardiac function with a consistent relationship between drug concentration and the duration of systolic ejection, a measure of the mechanism of action. Improvements in multiple measures of myocardial function, such as fractional shortening and stroke volume, have been demonstrated in all species, and in the dog, were associated with no change in myocardial oxygen consumption and thus an increase in myocardial efficiency. Finally, a few studies in healthy volunteers and patients with heart failure showed that CK-1827452 could safely increase cardiac performance with no evidence of arrhythmias or ischemia at therapeutic doses. For the greater than half a million patients who are admitted each year with heart failure and decreased LV systolic function, as well as the half million patients undergoing cardiac surgery and others requiring critical care inotropic support, the potential uses of a safe intravenous agent are large. In addition, for the millions of patients with chronic heart failure and reduced LV function, cardiac myosin activators offer the promise of a safe and effective treatment to address one of the underlying causes of heart failure. Phase III studies are being planned to test whether CK-1827452 will fulfill that promise.
References
Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y (2008) For the American heart association statistics committee and stroke statistics s. heart disease and stroke statistics-2008 Update: a report from the American heart association statistics Committee and stroke statistics subcommittee. Circulation 117:e25–e146. doi:10.1161/CIRCULATIONAHA.107.187998
Fonarow GC, Stough WG, Abraham WT, Albert NM, Gheorghiade M, Greenberg BH, O’Connor CM, Sun JL, Yancy CW, Young JB (2007) Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: a report from the OPTIMIZE-HF Registry. J Am Coll Cardiol 50:768–777. doi:10.1016/j.jacc.2007.04.064
Petersen JW, Felker GM (2008) Inotropes in the management of acute heart failure. Crit Care Med 36:S106–S111
Hinken AC, Solaro RJ (2007) A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda, MD) 22:73–80. doi:10.1152/physiol.00043.2006
Solaro RJ, de Tombe PP (2008) Review focus series: sarcomeric proteins as key elements in integrated control of cardiac function. Cardiovasc Res 77:616–618. doi:10.1093/cvr/cvn004
Spudich JA (1994) How molecular motors work. Nature 372:515–518. doi:10.1038/372515a0
Vale RD, Milligan RA (2000) The way things move: looking under the hood of molecular motor proteins. Science 288:88–95. doi:10.1126/science.288.5463.88
Cohn JN, Goldstein SO, Greenberg BH, Lorell BH, Bourge RC, Jaski BE, Gottlieb SO, McGrew F 3rd, DeMets DL, White BG (1998) A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. Vesnarinone trial investigators. N Engl J Med 339:1810–1816. doi:10.1056/NEJM199812173392503
Cuffe MS, Califf RM, Adams KF Jr, Benza R, Bourge R, Colucci WS, Massie BM, O’Connor CM, Pina I, Quigg R, Silver MA, Gheorghiade M (2002) Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. J Am Med Assoc 287:1541–1547. doi:10.1001/jama.287.12.1541
Felker GM, Benza RL, Chandler AB, Leimberger JD, Cuffe MS, Califf RM, Gheorghiade M, O’Connor CM (2003) Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol 41:997–1003. doi:10.1016/S0735-1097(02)02968-6
Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML et al (1991) Effect of oral milrinone on mortality in severe chronic heart failure. The promise study research group. N Engl J Med 325:1468–1475
Cytokinetics Inc. www.cytokinetics.com. Accessed 20 Oct 2008
Niu C, Cox DR, Lee KH, Sylvester S, Sueoka SH, Qian X, Feng B, Malik F, Morgans D, Hartman JJ, Sakowicz R, Elias KA (2004) Cellular responses of the myosin activator CK689705 in normal and heart failure. Mol Biol Cell 15:271A
Rodriguez HM, Sylvester S, Qian X, Morgan BP, Morgans DJ, Malik F, Sakowicz R (2004) Activation of cardiac sarcomere ATPase by CK1122534, a small molecule agent that specifically targets cardiac myosin. Mol Biol Cell 15:32A
Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ (2003) Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299:1743–1747. doi:10.1126/science.1081412
Niu CR, Anderson RL, Cox DR, Morgan BP, Morgans DJ, Malik F, Elias KA (2004) The cardiac myosin activator, CK1122534, increases contractility in adult cardiac myocytes but does not affect the calcium transient or depend on second messenger. Circulation 110:478–479
Malik F, Shen YT, Katori T, Sueoka SH, Anderson R, Cox D, Garard M, Hartman J, Kim SJ, Kraynack E, Kuklov A, Lee KH, Lu PP, Muci A, Niu C, Rodriguez H, Suehiro I, Sylvester S, Tochimoto T, Elias KA, Morgan BP, Sakowicz R, Kass DA, Vatner SF, Morgans DJ (2005) Direct activation of cardiac myosin, a novel mechanism for improving cardiac function. Circ Res 97:E34
Anderson RL, Sueoka SH, Rodriguez HM, Lee KH, Cox DR, Kawas R, Morgan BP, Sakowicz R, Morgans DJ, Malik F, Elias KA (2005) In vitro and in vivo efficacy of the cardiac myosin activator CK-1827452. Mol Bio Cell 16 (Abstract #1728)
Rodriguez HM, Kawas R, Hartman JJ, Malik F, Sakowicz R (2007) The cardiac myosin activator, CK-1827452, accelerates the enzymatic step gating entry of myosin into its force generating state. Biophysical society meeting abstracts. Biophys J 2007:481A
Anderson RL, Pokrovskii M, Elias KA (2007) Effects of cardiac myosin activators on excitation-contraction (E-C) coupling in venricular myocytes. Biophysical society meeting abstracts. Biophys J 2007:133a
Anderson RL, Sueoka SH, Lee KH, Rodriguez HM, Kawas RF, Godinez G, Morgan BP, Sakowicz R, Morgans DJ, Malik F, Elias KA (2006) In vitro and in vivo characterization of CK-1827452, a selective cardiac myosin activator. J Card Fail 12:S86. doi:10.1016/j.cardfail.2006.06.292
Shen YT, Zhang Y, Morgans DJ, Vatner SF and Malik F (2008) A novel inotropic agent that activates cardiac myosin and increases cardiac contractility without increasing MVO2 in heart failure with left ventricular hypertrophy. J Am Coll Card 51(10, Suppl 1):A54. doi:10.1016/j.jacc.2008.02.004
Teerlink JR, Malik FI, Clarke CP, Saikali KG, Escandon RD, Lee JH, Wolff AA (2006) The selective cardiac myosin activator, CK-1827452, increases left ventricular systolic function by increasing ejection time: results of a first-in-human study of a unique and novel mechanism. J Card Fail 12:763. doi:10.1016/j.cardfail.2006.10.007
Malik FI, Saikali KG, Clark CP, Teerlink JR, Wolff AA (2007) Systolic ejection time is a sensitive indicator of left ventricular systolic function during treatment with the selective cardiac myosin activator, CK-1827452. J Card Fail 13(6, Suppl 2):S145–S146. doi:10.1016/j.cardfail.2007.06.614
Malik F, Teerlink JR, Escandon RD, Clarke CP, Wolff AA (2006) The selective cardiac myosin activator, CK-1827452, a calcium-independent inotrope, increases left ventricular systolic function by increasing ejection time rather than the velocity of contraction. Circulation 114:II–441
Cleland JGF, Nifontov EM, McMurray JJV, Senior R, Lang CC, Clarke CP, Francis D, Greenberg BH, Mayet J, Monaghan M, Neyses L, Tsyrlin VA, Goldman JH, Teerlink JR, Brand G, Lee JH, Saikali KG, Wolff AA, Malik FI (2008) First clinical trial of the selective cardiac myosin activator, CK-1827452, in heart failure: effect of dose and plasma concentration on systolic function. Eur J Heart Fail Suppl 7:189–190. doi:10.1016/S1567-4215(08)60526-3
Cleland JGF, Malik FI (2008) The selective cardiac myosin activator, CK-1827452, increases systolic function in heart failure. J Card Fail 14:S67. doi:10.1016/j.cardfail.2008.10.012
Jerling M, Chew T, Escandon RD, Lee JH, Saikali KG, Venables E, Clarke CP, Malik F, Wolff AA (2007) Oral bioavailability of the selective cardiac myosin activator, CK-1827452: chronic oral inotropic therapy for heart failure? J Card Fail 13(6, Suppl 2):S148. doi:10.1016/j.cardfail.2007.06.622
Parker JD, Michaels AD, Kass DA, Wolff AA, Felker GM, Malik FI (2008) Rationale and design for a phase II study evaluating the effect of the cardiac myosin activator, CK-1827452, on cardiac function, hemodynamics, and myocardial oxygen consumption in patients with heart failure. J Card Fail 14:S79. doi:10.1016/j.cardfail.2008.06.438
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Teerlink, J.R. A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev 14, 289–298 (2009). https://doi.org/10.1007/s10741-009-9135-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-009-9135-0