
Abstract
Relaxin is a naturally occurring peptide hormone that plays a central role in the hemodynamic and renovascular adaptive changes that occur during pregnancy. Triggering similar changes could potentially be beneficial in the treatment of patients with heart failure. The effects of relaxin include the production of nitric oxide, inhibition of endothelin, inhibition of angiotensin II, production of VEGF, and production of matrix metalloproteinases. These effects lead to systemic and renal vasodilation, increased arterial compliance, and other vascular changes. The recognition of this has led to the study of relaxin for the treatment of heart failure. An initial pilot study has shown favorable hemodynamic effects in patients with heart failure, including reduction in ventricular filling pressures and increased cardiac output. The ongoing RELAX-AHF clinical program is designed to evaluate the effects of relaxin on the symptoms and outcomes in a large group of patients admitted to hospital for acute heart failure. This review will summarize both the biology of relaxin and the data supporting its potential efficacy in human heart failure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In 1926, Frederick Hisaw noted that serum from pregnant rabbits and guinea pigs injected into virgin guinea pigs resulted in relaxation of the pubic ligaments, and in 1930, a specific peptide was isolated and called relaxin [26, 37]. Over 40 years later, these same investigators, working with others, noted that relaxin had multiple other effects, including significant effects on vascular function [37]; reviewed by [24]. Since then, the cardiovascular and renal effects of relaxin have been investigated with a growing understanding of the molecular, cellular, physiologic, and clinical roles for this molecule (reviewed in [6, 24]).
The current understanding of relaxin indicates that it plays a central role in the hemodynamic and renovascular adaptive changes that occur during pregnancy (reviewed in [41]). Triggering similar changes could potentially be beneficial in the treatment of patients with heart failure. The recognition of this has led to the study of relaxin for this therapeutic application. The data available to date indicate that relaxin may be a “player in human heart failure” [21, 22]. This review will summarize both the biology of relaxin and the data supporting its potential efficacy in human heart failure.
Relaxin molecule
Relaxin, which is naturally produced by the corpus luteum during the menstrual cycle and pregnancy in women and by the prostate in men, is a 6-kDa peptide made up of 53 amino acids. The primary structure of this peptide is shown in Fig. 1. Relaxin has structural similarities to insulin, including its synthesis as a single-chain molecule that folds and undergoes removal of a C-peptide to yield a 2-chain molecule with conserved disulfide bonds. Relaxin and insulin, however, have distinctly different receptors and consistent with this, have non-overlapping biological properties.

Structure of native and manufactured human relaxin
Human relaxin has been manufactured and the synthesized molecule is identical in amino acid sequence to the mature, naturally occurring human relaxin.
Cardiovascular molecular biology of relaxin
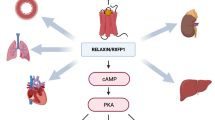
Recent advances have led to a preliminary understanding of the biology of relaxin receptors in the vasculature. Relaxin activity is initiated by binding to its cognate receptor, LGR7 (or RXFP1), a G-protein coupled receptor with a pKd of 9.26 [33]. LGR7 is a member of the leucine-rich repeats containing G-protein coupled receptor family (LGR) which are also known as relaxin family peptide receptors (RXFP) [38]. There is limited information on the location of relaxin receptors, but LGR7 is localized in small renal, mesenteric, and thoracic aortae in mice and rats of both sexes [48]. Relaxin binding sites have also been detected in blood vessels from humans, as well as in cells and tissues from the human heart [38]. LGR8 (or RXFP2), a receptor closely related to LGR7, has a binding affinity ~10-fold lower than LGR7, but may activate similar, but not identical, signaling pathways [35]. LGR8 receptors may be localized in tissues, including the vasculature, with a distribution pattern similar to that of LGR7 [38]. These data suggest that relaxin administered therapeutically could engage these vascular receptors to directly mediate hemodynamic changes.
Relaxin itself has also been detected in rat renal, mesenteric and thoracic aorta [48], and in human saphenous vein, mammary artery and vessels in the skin [42]. Therefore, a vessel-derived relaxin ligand-receptor system could also act locally to affect vascular function.
An important intracellular second messenger in relaxin signaling is cAMP (Fig. 2). Binding of relaxin to its receptor leads to activation of mitogen-activated protein kinase (MAPK) [23, 59], as well as phosphoinositide-3-kinase (PI3 K) [46], to induce increases in cAMP in target cells. In other cells in which responses to relaxin have been measured, only small, transient increases in cAMP [51] or MAPK pathway activation with no increases in cAMP [49] have been described, indicating the existence of alternate signaling pathways. Interestingly, signaling via the glucocorticoid receptor has also been described [23, 35].

Proposed mechanism of relaxin-mediated vasodilation and arterial compliance
Relaxin causes systemic and renal hemodynamic effects indirectly. Nitric oxide [2, 3, 12] and the endothelin type B receptor [7, 23] act as mediators of relaxin’s vasodilatory effect. Matrix metalloproteinases (MMPs), stimulated by relaxin, convert Big Endothelin to its bioactive form, Endothelin1–32, and are required for relaxin-mediated vasodilation [40]. Other potential mediators of relaxin’s vasodilatory activity are atrial natriuretic peptide [56], which is produced in the heart, and vascular endothelial growth factor (VEGF) in the endometrium [50, 58].
Recent evidence of “negative cooperativity” of relaxin binding kinetics [55] suggests that downstream events may occur with a biphasic (U- or bell-shaped) dose response. In concurrence, cAMP is induced with a bell-shaped dose response curve [34]. Further downstream, other effects that occur with biphasic dose response curves include stimulation of MMP expression by isolated cells [57], relaxin-induced systemic and renal hemodynamic responses in rats [13, 17, 18], and changes in osmolality in rats [13].
The stimulation of multiple vascular signaling pathways on the vascular system by relaxin may offer particular advantages in the treatment of heart failure. One of the possible limitations of prior vasodilator therapy has been that single mediators of heart failure have been specifically targeted, allowing induction of compensatory parallel pathways involving other neurohormones, leading to either loss of effect or the appearance of safety problems with increasing time or dose. Since relaxin works indirectly through multiple pathways with short- and long-term effects on hemodynamics, it may be particularly well suited for therapeutic treatment of heart failure, with acute and sustained effects, as well as a favorable benefit-risk profile.
Introduction to relaxin pharmacology
Naturally occurring relaxin possesses multiple systemic and renal vasodilatory properties that mediate maternal adjustments to the demands of pregnancy, as well as connective tissue remodeling, and angiogenic properties. During the first trimester, coincident with the rise in circulating relaxin levels, cardiac output and global arterial compliance both rise, systemic vascular resistance falls, and glomerular filtration rate (GFR) and renal blood flow (RBF) both increase. Nonclinical models and data from clinical trials indicate that relaxin is the circulating factor responsible for these changes. These data, as well as mechanisms of these actions, are summarized in the sections below.
Non-clinical pharmacology
In nonclinical pharmacology models, relaxin has been shown to cause systemic and renal vasodilation [44, 47] and increases in global arterial compliance.
Systemic vasodilation
In rat models, relaxin decreases systemic vascular resistance (SVR) (see Fig. 3); [8] and stimulates greater decreases in SVR, when baseline SVR is high [8]. The decrease in SVR is accompanied by an increase in cardiac output, primarily due to an increase in stroke volume [8]. Relaxin also inhibits the vasoconstrictive properties of two principal mediators of vasoconstriction in heart failure, endothelin and angiotensin II (see Fig. 4); [14, 15, 44].

Relaxin effect on systemic vascular resistance and cardiac output in rats (from [17])

Relaxin reverses the effects of angiotensin II on hemodynamic parameters in rats (adapted from [18])
Mesenteric vessels, as well as small renal vessels, from relaxin-treated rats showed reduced myogenic reactivity, compared with rats treated with vehicle [47]. Vessels from the mesenteric artery, but not the portal vein, isolated from relaxin-treated rats also showed blunted responses to vasoconstrictor agents, including norepinephrine and angiotensin II [44]. Relaxin has shown vasodilatory activity superior to acetylcholine or nitroprusside in isolated rat and guinea pig hearts [3]. Relaxin also induced vasodilation in isolated human resistance vessels when treated ex vivo [27].
Renal vasodilation
During pregnancy, vasodilation occurs in the renal vasculature causing maternal renal hemodynamic adjustments that anticipate the increasing metabolic demands of the fetus. GFR and RBF increase by ~45% during the first trimester, compared to pre-pregnant or post-partum values [16, 53]. Direct and indirect measurements on the kidney have concluded that vasodilation of both afferent and efferent renal arterioles occurs leading to no increase in glomerular capillary pressure [4]. In pregnancy, the increase in RBF exceeds that of the increase in GFR, leading to a decrease in calculated filtration fraction. The increase in GFR is believed to be mostly due to the increase in RBF. A large body of research in rodent models has suggested that these hemodynamic adjustments are, in large part, mediated by relaxin (reviewed in [39]).
The renal vasodilation that is observed during pregnancy has been reproduced following exogenous relaxin infusion in rats [14, 15]. GFR and effective renal plasma flow (ERPF) increased by 33% and 49% over baseline, respectively, in both male and ovariectomized female rats treated with relaxin and this effect occurred within 1–2 h of infusion of relaxin [13]. The relaxin-induced renal vasodilation and improved GFR were mediated by the endothelial ETB receptor subtype, presumably on endothelial cells [31] and nitric oxide (NO) [14, 15]. Because concurrent administration of inhibitors of NO and the ETB receptor to midterm pregnant rats produced no further decline in renal function than either agent given alone, a common vasodilatory pathway has been hypothesized [7]. Furthermore, because the results can be replicated using isolated renal arteries using myogenic reactivity as a bioassay for vasodilation, they indicate that relaxin acts directly on the vessel and that the signaling required, including induction of the MMPs, ET1–32 production leading to ETB receptor activation, and NO production, occur locally in the vessel (reviewed in [6]).
Arterial compliance
Activation of matrix metalloproteinases (MMPs) lead to increases in arterial compliance [15, 57]. Relaxin, infused for as short a period as 2–3 days in rats, has been shown to increase global arterial compliance, coincident with an increase in cardiac output and decrease in SVR [8] and consistent with its role in pregnancy [17]. The increase in compliance studied in an ex vivo model showed a relaxin-mediated increase in compliance, independent of smooth muscle-mediated changes in tone, suggesting that alterations in the extracellular matrix of the vessel wall contributed to increased compliance.
Prior human testing in non-heart failure conditions
Vasodilation in non-heart failure studies
Seven clinical trials evaluated the safety and efficacy of relaxin administered by continuous subcutaneous infusion at doses ranging from 6–200 μg/kg/day for periods of 6 months and up to 1 year in a few subjects (Data on file, Corthera Inc.). In these studies, 257 patients were treated with relaxin and 124 patients received placebo. Relaxin treatment of systemic sclerosis patients was associated with statistically significant decreases, within normal limits, of systolic and diastolic blood pressure in clinical trials. The decrease in systolic blood pressure was greater by ~two-fold in subjects, who were hypertensive (SBP > 140 mm Hg) upon entry into the trial, compared to the group as a whole (Fig. 5). No cases of symptomatic hypotension were reported in relaxin-treated subjects in any of the systemic sclerosis trials.

Effect of relaxin on systolic blood pressure in normotensive and hypertensive human subjects
Renal function in healthy volunteers
In a physician-sponsored open-label research study of renal function in normal volunteers conducted in Great Britain, 11 healthy male and female subjects were dosed with an IV infusion of relaxin at 0.5–2.0 μg/kg/h, targeting pregnancy serum concentrations, for 5 h [54]. GFR and RBF were measured using inulin and PAH, respectively, according to established methods. RBF increased by 43% but an increase in GFR was not detected (see Table 1). The finding that GFR was unaffected by relaxin is at odds with nonclinical and clinical data indicating that relaxin increases GFR. It is possible that in this particular study, in healthy volunteers, the dose of relaxin, the short period of infusion, the volume status of the subjects, renal autoregulatory responses present in these normal subjects, or all of those, played a role in circumventing a concurrent increase in GFR. Relaxin also showed a modest natriuretic effect in this study.
Renal function in non-heart failure studies
In the systemic sclerosis studies described above, relaxin administration to subjects with systemic sclerosis was associated with statistically significant increases in predicted creatinine clearance over the 6 month course of infusion (see Fig. 6); [25]; Data on file, Corthera Inc.). In support of these results, serum creatinine, uric acid and BUN levels were also decreased in these subjects. These findings suggest that relaxin mediates increases in renal function in humans.

Effect of relaxin on predicted creatinine clearance in human subjects
Relaxin role in acute heart failure
The demonstrated effects of relaxin suggest that it may be therapeutic in clinical settings where reductions in systemic vascular resistance and improved renal blood flow may be beneficial. The ADHERE and OPTIMIZE registries have confirmed on a large scale earlier observations that a substantially higher proportion of acute heart failure events than was previously recognized are associated with elevated blood pressure, most probably induced by vasoconstriction and vasomotor nephropathy, or renal vasoconstriction [1, 9, 32]. These symptoms may be mediated by abnormal activation of the Renin-Angiotensin-Aldosterone System (RAAS), as well as of endothelin. These patients represent the majority of patients with acute heart failure and in some recent classifications have been designated as having acute vascular failure or acute cardiovascular (hypertensive) failure, in contrast to those with acute decompensated (cardiac) failure [1, 10, 11, 45].
In addition, decreased renal blood flow has been recognized as a major contributing factor to the renal dysfunction that often accompanies acute heart failure [29, 30, 43]. The degree of renal dysfunction is an important prognostic factor for these patients [19, 52].
Potential mediators of this clinical syndrome include activation of the RAAS, sympathetic nervous system, and endothelin systems, as well as resistance to or failure of counter-regulatory systems such as the natriuretic peptides [9, 32].
In numerous nonclinical studies, relaxin has demonstrated the ability to prevent or reverse the effects of the neurohormones angiotensin II and endothelin. Acute infusion of relaxin restored cardiac output, systemic vascular resistance, and arterial compliance that were dysregulated by infusion of angiotensin II in rats [18]. In isolated rat aorta and mesenteric artery, relaxin pretreatment mitigated the maximum contractile response to endothelin-1 by ~40% [23]. In rat studies, renal vasoconstriction induced by angiotensin II, measured as decreases in GFR and RBF, was reversed by infusion of relaxin [14]. In vitro, relaxin also antagonized the effect of angiotensin II on increased collagen deposition and proliferation in rat cardiac fibroblasts [51], potentially by inhibition of the Smad transcription factors required for extracellular matrix synthesis [36].
Gene expression for relaxin and the relaxin receptor has been detected in rat renal, mesenteric and thoracic aorta [48], and in human saphenous vein, mammary artery, and vessels in the skin [42]. These data suggest that there is likely a vessel-derived relaxin ligand-receptor system that can act locally to affect vascular function in humans. Perhaps more importantly, the presence of relaxin receptors in blood vessels strongly suggests that relaxin can engage these receptors when given therapeutically. Relaxin’s vasodilatory activity has been attributed to NO acting as the ultimate effector molecule [2, 14, 23], and it is also likely that vasodilation is mediated via the endothelial endothelin type B receptor.
Relaxin gene expression has been shown to be upregulated in atria and ventricles from patients with heart failure, compared to normal subjects [21, 22]. Elevated left ventricular end-diastolic pressure in isolated rat hearts stimulated relaxin gene expression, providing a possible mechanism for the finding of enhanced relaxin expression in ventricles from heart failure patients. These increases may be reflected in increased serum relaxin in patients with compensated heart failure [21, 22, 28]. These elevated levels, though modest (8–16 pg/ml and 11–644 pg/ml in CHF patients in the two cited studies; ≤2 pg/ml in control subjects), compared to the 1000 pg/ml levels attained in pregnant women, suggest compensatory upregulation of relaxin in patients with heart failure. Relaxin did not appear to be prognostic for outcome [28].
Since relaxin can decrease SVR, allowing increased cardiac output, and can also improve or preserve renal perfusion, it may be ideally suited for the treatment of patients with heart failure, and in particular, patients with acute vascular failure. In addition, relaxin-mediated increases in arterial compliance, in conjunction with vasodilation, may lead to improvements in both steady and pulsatile arterial load components of arterial mechanics [39], making it a unique potential therapeutic in heart failure.
Phase 1 heart failure study
Relaxin in compensated CHF
A single site, open-label study of relaxin in patients with compensated CHF has been completed at the Charite Hospital in Germany [20]. This was a safety and dose-finding study of intravenous relaxin given for 24 h at doses ranging from 10 to 960 μg/kg/day. Pharmacodynamic dose-response parameters (serial hemodynamic measurements using pulmonary artery and radial artery catheters and serial renal chemistry parameters) were evaluated to define relaxin doses for further study.
The study enrolled 16 subjects with compensated CHF, NYHA Class II–III due to ischemic heart disease, hypertensive heart disease, or dilated cardiomyopathy with left ventricular ejection fraction <35%, pulmonary capillary wedge pressure (PCWP) ≥ 16 mmHg and cardiac index (CI) ≤ 2.5 l/min/m2. All 16 subjects completed dosing and the Day 9 follow-up visit. Relaxin was safe and well-tolerated in all subjects.
Trends in improvement in a number of hemodynamic parameters were observed during dosing and for several hours thereafter. Doses of relaxin in the range of 10–100 μg/kg/day appeared to have a more pronounced effect than higher doses on right atrial pressure, pulmonary artery pressure, PCWP and NT-pro-BNP, while higher doses in the range of 240–960 μg/kg/day tended to have a greater effect on CI (see Fig. 7). Values for systemic vascular resistance decreased at all doses. No consistent changes in heart rate or blood pressure were observed either during or post-dosing.
The different dose responses observed for the hemodynamic parameters may be explained by imbalances in baseline hemodynamic status of the dose groups or may be due to random variability in a small pilot study. However, certain biological effects of relaxin are known to follow a U-shaped dose-response curve [13, 18, 55, 57]. Alternatively, counter-regulatory neurohormonal mechanisms may have been stimulated at higher relaxin doses, thus over-powering the salutary responses observed at the lower doses. An intriguing alternative explanation may be that doses in the lower range produce more venous vasodilation, (decrease in PCWP without change in CO/CI) while higher doses may produce more arterial vasodilation. Additional well-controlled hemodynamic studies will be required to provide further insight into this pattern.

Pulmonary capillary wedge pressure and cardiac index-infusion (black bars) and post-infusion (white bars) (24 h each). Vertical lines mark dosage increases every 8 h (all in μg/kg/day). *P < 0.05 vs. baseline (Data courtesy [20])
Serum markers of renal function reflected the improvement in kidney function mediated by relaxin, i.e., decreases in creatinine, uric acid, and BUN were observed during dosing, presumably due to renal vasodilation. Small increases in creatinine and BUN above baseline were observed transiently at Day 9 post-dosing in the 960 μg/kg/day relaxin dose group. Although these increases were not associated with clinical significance, it is possible that these observations may represent nascent dose limiting toxicity due to counter-regulatory mechanisms (rebound). Hence, the dose of 960 μg/kg/day was regarded as the maximally tolerated dose of IV relaxin in these patients with heart failure.
Phase 2/3 therapeutic heart failure studies
As noted above, a substantially higher proportion of patients with acute heart failure than was previously recognized have elevated blood pressure at the time of presentation, so called acute vascular failure. Renal dysfunction is a common co-morbidity and major predictor of poor outcomes in all patients with acute heart failure, and appears to be particularly common in patients with acute vascular failure compared to those with other presentations [5]. As of yet, no therapy has been demonstrated conclusively to improve symptoms or renal function in this group of patients. The current understanding of the hemodynamic and renovascular effects of relaxin, as well as the encouraging results from the evaluation of relaxin in patients with compensated heart failure, supported the investigation of the use of relaxin as a therapeutic agent for the treatment of patients with acute vascular failure. The RELAX-AHF study has been undertaken to evaluate the effects of relaxin therapy on symptoms, signs, and outcomes in these patients.
RELAX-AHF is a multicenter, placebo-controlled, double-blind, randomized, international trial assessing the effects of intravenous relaxin in patients hospitalized with acute heart failure (AHF). The RELAX-AHF trial includes patients aged >18 years, hospitalized for AHF (defined as moderate or marked dyspnea, pulmonary congestion on chest x-ray and elevated BNP), baseline systolic blood pressure >125 mmHg, impaired renal function (estimated creatinine clearance of 30–75 ml/min/1.73 m2 by simplified Modification of Diet in Renal Disease formula). Eligible patients will have received intravenous furosemide and are randomized within 16 h of presentation. Endpoints will include serial assessment of dyspnea using Visual Analog Scale and 7-point Likert Scale. In addition, in-hospital medication use, heart failure signs and symptoms, weight loss, incidence of worsening heart failure, and the length of hospital stay will be assessed. Serum creatinine will be assessed serially until Day 14 to assess renal function. After discharge and until Day 180, mortality and cardiorenal rehospitalizations will be recorded.
A multicenter, international pilot study, Pre-RELAX-AHF, has recently been conducted to determine the optimal dose of relaxin to test in the main study, RELAX-AHF. A total of 234 patients were randomized in the pilot study to receive intravenous placebo or relaxin at doses of 10, 30, 100, and 250 ug/kg/d for 48 h.
Future directions
The naturally occurring peptide hormone relaxin holds promise as a novel pleiotropic vasodilator for the treatment of patients with acute heart failure and preserved or elevated blood pressure (acute vascular failure). The effects of relaxin include the production of nitric oxide, inhibition of endothelin, inhibition of angiotensin II, production of VEGF, and production of MMPs. These multiple mechanisms by which relaxin causes systemic and renal vasodilation, increased arterial compliance and other vascular changes may prove beneficial in these patients.
References
Adams KF, Fonarow GC et al (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100, 000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 149:209–216. doi:10.1016/j.ahj.2004.08.005
Bani D, Failli P et al (1998) Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension 31(6):1240–1247
Bani-Sacchi T, Bigazzi M et al (1995) Relaxin-induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol 116(1):1589–1594
Baylis C (1999) Relaxin may be the “elusive” renal vasodilatory agent of normal pregnancy. Am J Kidney Dis 34(6): 1142–1144; discussion 1144–1145
Butler J, Forman DE et al (2004) Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J 147(2):331–338
Conrad KP, Novak J (2004) Emerging role of relaxin in renal and cardiovascular function. Am J Physiol Regul Integr Comp Physiol 287:R250–R261. doi:10.1152/ajpregu.00672.2003
Conrad KP, Gandley RE et al (1999) Endothelin mediates renal vasodilation and hyperfiltration during pregnancy in chronically instrumented conscious rats. Am J Physiol 276(5 Pt 2):F767–F776
Conrad KP, Debrah DO et al (2004) Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 145(7):3289–3296. doi:10.1210/en.2003-1612
Cotter G et al (2002) Acute heart failure: a novel approach to its pathogenesis and treatment. Eur J Heart Fail 4:227–234. doi:10.1016/S1388-9842(02)00017-X
Cotter G, Felker GM et al (2008) The pathophysiology of acute heart failure—Is it all about fluid accumulation? Am Heart J 155:9–18. doi:10.1016/j.ahj.2006.02.038
Cotter G, Metra M et al (2008) Fluid overload in acute heart failure—Re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail 10:165–169. doi:10.1016/j.ejheart.2008.01.007
Danielson LA, Conrad KP (1995) Acute blockade of nitric oxide synthase inhibits renal vasodilation and hyperfiltration during pregnancy in chronically instrumented conscious rats. J Clin Invest 96(1):482–490. doi:10.1172/JCI118059
Danielson LA, Conrad KP (2003) Time course and dose response of relaxin-mediated renal vasodilation, hyperfiltration, and changes in plasma osmolality in conscious rats. J Appl Physiol 95:1509–1514
Danielson LA, Sherwood OD et al (1999) Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest 103(4):525–533. doi:10.1172/JCI5630
Danielson LA, Kercher LJ et al (2000) Impact of gender and endothelin on renal vasodilation and hyperfiltration induced by relaxin in conscious rats. Am J Physiol Regul Integr Comp Physiol 279(4):R1298–R1304
Davison JM, Noble MCB (1981) Serial changes in 24-hour creatinine clearance during normal menstrual cycles and the first trimester of pregnancy. Br J Obstet Gynecol 88:10–17
Debrah DO, Conrad KP et al (2005) Effects of relaxin on systemic arterial hemodynamics and mechanical properties in conscious rats: sex dependency and dose response. J Appl Physiol 98:1013–1020
Debrah DO, Conrad KP et al (2005) Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46(4):745–750. doi:10.1161/01.HYP.0000184230.52059.33
Dries D, Exner D, Domanski M, Greenberg B, Stevenson L (2000) The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dusfunction. J Am Coll Cardiol 35:681–689. doi:10.1016/S0735-1097(99)00608-7
Dschietzig T, Unemori E et al (2009) A pilot safety, tolerability and pharmacodynamic trial of intravenous recombinant human relaxin in compensated heart failure. J Card Fail (in press)
Dschietzig T, Richter C et al (2001) Flow-induced pressure differentially regulates endothelin-1, urotensin II, adrenomedullin, and relaxin in pulmonary vascular endothelium. Biochem Biophys Res Commun 289(1):245–251. doi:10.1006/bbrc.2001.5946
Dschietzig T, Richter C et al (2001) The pregnancy hormone relaxin is a player in human heart failure. FASEB J 15(12):2187–2195. doi:10.1096/fj.01-0070com
Dschietzig T, Bartsch C et al (2003) Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-kappaB. Circ Res 92(1):32–40. doi:10.1161/01.RES.0000051884.27117.7E
Dschietzig T, Bartsch C et al (2006) Relaxin-A pleiotropic hormone and its emerging role for experimental and clinical therapeutics. Pharmacol Ther 112(1):38–56. doi:10.1016/j.pharmthera.2006.03.004
Erikson MS, Unemori E (2001) Relaxin clinical trials in systemic sclerosis. Relaxin 2000. G. Tregear, R Ivell, R Bathgate, JD Wade. Kluwer Academic Publishers, Dordrecht, pp 373–381
Fevold HL, Hisaw FL, Meyer RK (1930) The relaxative hormone of the corpus luteum. Its purification and concentration. J Am Chem Soc 52:3340–3348. doi:10.1021/ja01371a051
Fisher C, MacLean M et al (2002) Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation 106(3):292–295. doi:10.1161/01.CIR.0000025630.05387.45
Fisher C, Berry C et al (2003) N-terminal pro B type natriuretic peptide, but not the new putative cardiac hormone relaxin, predicts prognosis in patients with chronic heart failure. Heart (British Cardiac Society) 89(8):879–881. doi:10.1136/heart.89.8.879
Fonarow GC, Heywood TJ (2006) The confounding issue of comorbid renal insufficiency. Am J Med 119:S17–S25. doi:10.1016/j.amjmed.2006.09.013
Francis G (2006) Acute decompensated heart failure: the cardiorenal syndrome. Cleve Clin J Med 73(suppl 2):S8–S13
Gellai M, DeWolf R et al (1997) Contribution of endogenous endothelin-1 to the maintenance of vascular tone: role of nitric oxide. Pharmacology 55:299–308. doi:10.1159/000139542
Gheorghiade M, Abraham WT et al (2006) Systolic blood pressure at admission, clinical characteristics, and outcomes in patients hospitalized with acute heart failure. J Am Med Assoc 296(18):2217–2226. doi:10.1001/jama.296.18.2217
Halls ML, Bond CP et al (2005) Multiple binding sites revealed by interaction of relaxin family peptides with native and chimeric relaxin family peptide receptors 1 and 2 (LGR7 and LGR8). J Pharmacol Exp Ther 313(2):677–687. doi:10.1124/jpet.104.080655
Halls ML, Bathgate RA et al (2006) Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP Signaling by distinct mechanisms. Mol Pharmacol 70:214–226
Halls ML, Bathgate RA et al (2007) Comparison of signaling pathways activated by the relaxin family peptide receptors, RXFP1 and RXFP2, using reporter genes. J Pharmacol Exp Ther 320:281–290. doi:10.1124/jpet.106.113225
Heeg MH, Koziolek MJ et al (2005) The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int 68:96–109. doi:10.1111/j.1523-1755.2005.00384.x
Hisaw FL, Hisaw FL Jr, Dawson AB (1967) Effects of relaxin on the endothelium of endometrial blood vessels in monkeys (Macaca mulatta). Endocrinology 81:375–385
Hsu SY, Nakabayashi K et al (2002) Activation of orphan receptors by the hormone relaxin. Science 295(5555):671–674. doi:10.1126/science.1065654
Jeyabalan A, Conrad KP (2007) Renal function during normal pregnancy and preeclampsia. Front Biosci 12:2425–2437. doi:10.2741/2244
Jeyabalan A, Novak J et al (2003) Essential role for vascular gelatinase activity in relaxin-induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small renal arteries. Circ Res 93:1249–1257. doi:10.1161/01.RES.0000104086.43830.6C
Jeyabalan A, Shroff SG et al (2007) The vascular actions of relaxin. Adv Exp Med Biol 612:65–87
Kohsaka T, Min G et al (1998) Identification of specific relaxin-binding cells in the human female. Biol Reprod 59(4):991–999. doi:10.1095/biolreprod59.4.991
Liang KV, Williams AW, Greene EL, Redfield MM (2008) Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med 36:S75–S88
Massicotte G, Parent A et al (1989) Blunted responses to vasoconstrictors in mesenteric vasculature but not in portal vein of spontaneously hypertensive rats treated with relaxin. Proc Soc Exp Biol Med 190(3): 254–259
Milo-Cotter O, Adams KF et al (2007) Acute heart failure associated with high admission blood pressure—A distinct vascular disorder? Eur J Heart Fail 9:178–183. doi:10.1016/j.ejheart.2006.06.004
Nguyen BT, Yang L et al (2003) Phosphoinositide 3-Kinase activity is required for biphasic stimulation of cyclic AMP by relaxin. Mol Endocrinol (Baltimore, Md) 17:1075–1084. doi:10.1210/me.2002-0284
Novak J, Ramirez RJ et al (2002) Myogenic reactivity is reduced in small renal arteries isolated from relaxin-treated rats. Am J Physiol Regul Integr Comp Physiol 283(2):R349–R355
Novak J, Parry LJ et al (2006) Evidence for local relaxin ligand-receptor expression and function in arteries. FASEB J 20(13):2352–2362. doi:10.1096/fj.06-6263com
Palejwala S, Stein D et al (1998) Demonstration of a relaxin receptor and relaxin-stimulated tyrosine phosphorylation in human lower uterine segment fibroblasts. Endocrinology 139(3):1208–1212. doi:10.1210/en.139.3.1208
Palejwala S, Tseng L et al (2002) Relaxin gene and protein expression and its regulation of procollagenase and vascular endothelial growth factor in human endometrial cells. Biol Reprod 66(6):1743–1748. doi:10.1095/biolreprod66.6.1743
Samuel CS, Unemori EN et al (2004) Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 145(9):4125–4133. doi:10.1210/en.2004-0209
Schrier RW (2006) Role of diminished renal function in cardiovascular mortality: marker of pathogenetic factor? J Am Coll Cardiol 47:1–8. doi:10.1016/j.jacc.2005.07.067
Schrier RW, Dürr JA (1987) Pregnancy: an overfill or underfill state. Am J Kidney Dis 9(4):284–289
Smith MC, Danielson LA, Conrad KP et al (2006) Influence of recombinant human relaxin on renal hemodynamics in healthy volunteers. J Am Soc Nephrol 17:3192–3197. doi:10.1681/ASN.2005090950
Svendsen AM, Zalesko A et al (2008) Negative cooperativity in H2 relaxin binding to a dimeric relaxin family peptide receptor 1. Mol cell Endocrinol 296(1–2):10–17
Toth M, Taskinen P et al (1996) Relaxin stimulates atrial natriuretic peptide secretion in perfused rat heart. J Endocrinol 150(3):487–495. doi:10.1677/joe.0.1500487
Unemori EN, Amento EP (1990) Relaxin modulates synthesis and secretion of procollagenase and collagen by human dermal fibroblasts. J Biol Chem 265(18):10681–10685
Unemori EN, Erikson ME et al (1999) Relaxin stimulates expression of vascular endothelial growth factor in normal human endometrial cells in vitro and is associated with menometrorrhagia in women. Hum Reprod (Oxford, England) 14(3):800–806. doi:10.1093/humrep/14.3.800
Zhang Q, Liu SH et al (2002) Relaxin activates the MAP kinase pathway in human endometrial stromal cells. J Cell Biochem 85(3):536–544. doi:10.1002/jcb.10150
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Teichman, S.L., Unemori, E., Dschietzig, T. et al. Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail Rev 14, 321–329 (2009). https://doi.org/10.1007/s10741-008-9129-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-008-9129-3