
Abstract
Oryza sativa SPL14 (OsSPL14), identified as the IDEAL PLANT ARCHITECTURE1 or WEALTHY FARMER’S PANICLE gene, plays a critical role in regulating rice plant architecture. Here, OsSPL14-overexpression transgenic rice plants had shorter growth periods, short narrow flag leaves, and thick green leaves compared with wild type ‘MH86’ plants (WT). Additionally, transgenic lines had higher chlorophyll a (Chl a), chlorophyll b (Chl b), and carotenoid (Car x) contents at both seedling and mature stages. Expression of OsSPL14 increased at transcriptional level, and OsSPL14 protein level was substantially increased in transgenic lines relative to WT. A transcriptome analysis identified 473 up-regulated and 103 down-regulated genes in the transgenic plants. The expression of differentially expressed genes (DEGs) involved in carotenoid biosynthesis, abscisic acid (ABA) metabolism, and lignin biosynthesis increased significantly. Most of DEGs participated in “plant hormone signal transduction” and “starch and sucrose metabolism” were also up-regulated in the transgenic plants. In addition, there were higher ABA and gibberellin acid 3 (GA3) levels in OsSPL14-overexpression rice plants at seedling and tillering stages compared with WT. In contrast with that of WT, lignin and cellulose contents of culm increased distinctly. Also, silicon and potassium contents increased dramatically in transgenic lines. Meanwhile, the chalkiness ratios and chalkiness degrees decreased, and the gel consistency levels improved in transgenic lines. Thus, overexpression of OsSPL14 influenced growth period, leaf development, hormonal levels, culm composition, and grain quality characters of rice, which provides more insight into the function of OsSPL14.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa) SPL14 is a member of the SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) genes. SPL genes encode plant-specific transcription factors that have a highly conserved zinc ion-containing DNA-binding domain, known as a SQUAMOSA PROMOTER BINDING PROTEIN [SBP]-box (Yamasaki et al. 2004). SPB1 and SPB2 were the original SPL genes identified in Antirrhinum majus, and they bind to the promoter of the floral meristem identity gene SQUAMOSA (Klein et al. 1996).
Many SPL genes contain microRNA (miRNA) target sites, including those of miR156/157, and the target sites are located in the coding regions or 3′ untranslated regions (UTRs). In Arabidopsis thaliana, 10 of 16 SPL genes are predicted to be targets of miR156 (Rhoades et al. 2002; Schwab et al. 2005). SPL3, -4, and -5 each contain a target site for miR156 in their 3′ UTRs and are strongly repressed by miR156. Overexpression of SPL3 accelerates flowering, and plants expressing SPL3 having a mutated or lacking miR156 target site appeared earlier flowering and fewer leaves (Cardon et al. 1997; Wu and Poethig 2006; Gandikota et al. 2007). Additionally, paralogous genes SPL9 and SPL15, having miR156 target sites, are involved in controlling the juvenile-to-adult phase transition, and spl9 spl15 double mutants have shortened plastochrons, altered inflorescence architecture, and enhanced branching (Schwarz et al. 2008; Wang et al. 2008). SPL8, which contains no miRNA target sites, affects megasporogenesis, trichome formation on sepals, and stamen filament elongation (Unte et al. 2003). Accordingly, a series of studies on SPL genes in A. thaliana suggested that AtSPL genes are mainly associated with plant development and flowering.
In rice, there are 19 SPL genes that are unevenly distributed in the genome. Among them, 11 OsSPL genes are putative targets of OsmiR156. OsSPL genes are expressed in various tissues, including root, leaf, stem, panicle, stamen, and pistil, with most of the genes being predominantly expressed in the young panicles (Xie et al. 2006). OsSPL8, participating in building the laminar joints between leaf blades and leaf sheath boundaries, controls the development of ligules and auricles (Lee et al. 2007). OsSPL7, a target of OsmiR156, regulates tiller number and plant height. OsSPL7 overexpressing plants showed reduced tiller number, while OsSPL7 RNAi plants showed increased tiller number and reduced height (Dai et al. 2018). OsSPL3, another target of OsmiR156, regulates crow root development, and mutant of OsSPL3 had fewer crown roots (Shao et al. 2019). OsSPL14, also known as the IDEAL PLANT ARCHITECTURE1 (IPA1) or WEALTHY FARMER’S PANICLE (WFP) gene, is a homolog of Arabidopsis SPL9/SPL15, and it is regulated by OsmiR156/OsmiR529 (Jeong et al. 2011; Yue et al. 2017). A point mutation in the target site for OsmiR156 resulted in a significantly increased OsSPL14 protein level, which generated the mutation with reduced tiller number, increased grain number and grain weight, produced stout stems and improved lodging resistance (Jiao et al. 2010). An epigenetic change in the OsSPL14 promoter resulted in the same phenotype (Miura et al. 2010). Introduction of the functionally epigenetic OsSPL14WFP allele into elite indica rice genomes resulted in greatly improved panicle traits and grain yield (Kim et al. 2018). Moreover, the three tandem repeats in the upstream regions of IPA1/OsSPL14 enhanced the open chromatin configuration and reduced DNA methylation at the IPA1/OsSPL14 promoter, resulting in the optimal OsSPL14 expression level and ideal plant architecture, having the proper tiller number and large panicles (Zhang et al. 2017). Furthermore, the ipa1 loss-of-function mutants generated by CRISPR/Cas9 had a dwarf phenotype with increased tiller numbers (Li et al. 2016). A genome-wide expression profiling analysis suggested that IPA1/OsSPL14 could directly bind to the SBP-box target motif GTAC and regulate the expression of TEOSINTE BRANCHED1 (TB1) and DENSE AND ERECTPANICLE1 (DEP1), which influence tiller number, plant height, and panicle length (Lu et al. 2013). Additionally, higher IPA1 levels can enhance immunity in rice. Phosphorylated IPA1/OsSPL14 activates the expression of WRKY45, which then enhances blast disease resistance (Wang et al. 2018). Overexpression of IPA1/OsSPL14 also enhances disease resistance against bacterial blight (Liu et al. 2019). Because of an important role of IPA1/OsSPL14 in rice, studies on the regulatory mechanisms influencing its expression were carried out. A RING-finger E3 ligase, IPA1 INTERACTING PROTEIN1 (IPI1), could regulate the protein levels of IPA1/OsSPL14 in different tissues (Wang et al. 2017a). A human ovarian tumor domain-containing ubiquitin aldehyde-binding protein 1 like deubiquitinating enzyme in rice could promote the degradation of IPA1/OsSPL14 indirectly (Wang et al. 2017b). DWARF 53 (D53), a key repressor of the strigolactone (SL) signaling pathway, suppresses the transcription of IPA1/OsSPL14 (Song et al. 2017). SHORT INTERNODE1 (OsSHI1), by interacting with IPA1/OsSPL14, reduces the DNA-binding ability of IPA1/OsSPL14 to modulate the expression of downstream target genes (Duan et al. 2019). In short, IPA1/OsSPL14 plays important roles in controlling plant architecture and participates in disease resistance. Also, gene regulatory networks related to IPA1/OsSPL14 are complex and the corresponding molecular mechanisms require further clarification.
In this study, we introduced OsSPL14 into the indica cultivar ‘MH86′ and obtained transgenic lines that showed shorter growth periods, short narrow flag leaves, fewer tillers, and strong culms compared with the non-transgenic parental line. A transcriptome analysis indicated that genes involved in carotenoid biosynthesis and lignin biosynthesis were up-regulated in transgenic lines. The levels of ABA and GA3 increased, and culm lignin, cellulose, silicon, and potassium contents also increased. These results supplement the functional description of OsSPL14.
Materials and methods
Generation of transgenic rice
The plasmid pCAMBIA1300-OsSPL14 (Supplementary Fig. S1; OsSPL14/IPA1: GenBank GU136674.1; 7229 bp containing the promoter and coding region for mRNA) was introduced into mature rice (O. sativa L. indica cultivar ‘MH86’) embryos using Agrobacterium-mediated transformation. The hygromycin resistant plants were detected by PCR, RT-PCR and southern blot, then positive plants were grown in transgenic experimental field. Stably inherited transgenic plants possessing single copy insertions of the transgene were selected and used in this study.
RNA sequencing and transcriptome analysis
Total RNA was isolated from 1-month-old seedlings of transgenic plants and ‘MH86’ using TRIzol reagent. The cDNA library was constructed and sequenced using an Illumina HiSeq2500 Sequencer. Low quality reads were removed to produce clean reads, then the clean reads were mapped to the reference genome (https://plants.ensembl.org/Oryza_indica/Info/Index) using TopHat2 (https://ccb.jhu.edu/software/tophat.) to obtain mapped reads. Based on location information of mapped reads, gene expression levels were calculated by cuffquant and cuffnorm components of cufflinks software using fragments per kilobase of transcript per million fragments mapped (FPKM) as measurement index. According to screening standard of fold Change (FC) ≥ 2 and false discovery rate (FDR) < 0.01, DEGs analysis between transgenic plants and WT was perform by a differential expression analysis for sequence count data. Gene ontology (GO) enrichment analysis of DEGs was implemented by biological annotation system of the GO Consortium. The most enriched GO terms were conducted by KOBAS (2.0) with FDR ≤ 0.05. And the DEGs were mapped in Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Quantitative real-time PCR (qRT-PCR)
To validate DEGs identified in the transcriptome analysis and to analyze some genes from OsSPL14 regulatory networks, qRT-PCR was conducted on selected genes. The qRT-PCR was performed on a 7500 real-time PCR system (Applied Biosystems) with the Fast Start Universal SYBR Green Master system (Roche, USA), following the manufacturers’ protocols. The relative quantitative method (ΔΔCT) was used to evaluate the quantitative variation in the examined replicates. Primers were listed in Supplementary Table S1. TB F/R and DEP F/R primers, D53 F/R primers, and D17 F/R, D14 F/R, D3 F/R primers were those of Lu et al. (2013), Song et al. (2017), and Sun et al. (2014), respectively.
Determination of OsSPL14 protein abundance by western blotting
Total protein was extracted with buffer (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 4 mM MgCl2, 0.5% NP-40 detergent, and 5 mM DTT) and quantified using a modified Bradford protein assay kit (Sangon Biotech). Protein samples were separated in a 10% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. Immunodetection of OsSPL14 was performed using an anti-OsSPL14 primary antibody (1:1000) and a secondary antibody (1:10,000). HSP, as the endogenous control, was detected using anti-HSP primary antibody (1:10,000) (Beijing Protein Innovation) and secondary antibody (1:10,000). The signal was detected using a SuperSignal ECL chemiluminescence kit (Boster Biological Technology Co. Ltd).
ABA, jasmonate acid (JA), and GA3 assays
One-month-old seedlings and tillering-stage plants of the transgenic lines and WT were collected. ABA, JA, and GA3 contents were detected by high-performance liquid chromatography (HPLC, Agilent 1100).
Scanning electron microscopy (SEM) analysis
At the heading stage, a 0.3-cm section in the middle of the third internode was removed using a scalpel and placed in 3% glutaraldehyde for fixation for 8–10 h. After a series of treatment, the samples were mounted onto aluminum stubs, coated with carbon gold and were observed using a 3500 N scanning electron microscope (Hitachi, Tokyo, Japan) to collect images.
Compositional analysis of culm
At the mature stage, plant culm was collected and placed in an oven for 30 min at 105 °C and then transferred to 65 °C until a constant weight was achieved. The determination of cellulose, lignin and potassium (K) content were performed in accordance with the methods described in GB/T 13885-2003/ISO 6869:2000; (2000). The silicon (Si) content was determined using the Si molybdenum blue colorimetric method (Tong et al. 2005; Dai et al. 2005).
Statistical analyses
Statistical analyses were performed using one-way anova or Student’s t tests. P-values of < 0.05 were considered to indicate statistical significance. Statistical calculations were performed using Microsoft Excel 2016.
Results
Phenotype of MH86 over-expressing OsSPL14
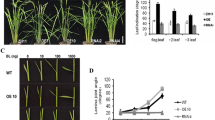
We introduced pCAMBIA1300-OsSPL14 into indica cultivar MH86 using Agrobacterium-mediated transformation and generated the OsSPL14-overexpression rice plants (OE; Supplementary Fig. S2). Two individual transgenic lines with single copy insertions, named as OE line 1 and OE line 2, were randomly selected for further study. In contrast, the growth periods of the OsSPL14 OE lines were shortened by 5–15 d, depending on the growing area (Fig. 1a). The expression of OsSPL14 increased in OE lines, and immunoblot analysis validated that OsSPL14 protein level was substantially increased (Fig. 1c, d). OE lines had shorter and narrower flag leaves compared with those of WT plants. The flag leaf lengths and widths of OE plants decreasing by about one third and one quarter, respectively (Fig. 1b, e, f). Consistent with a previous report (Jiao et al. 2010), OE plants showed decreased tiller numbers and strong culms (Fig. 1 g). The culm diameters and culm wall thicknesses of OE lines increased by ~ 21–24% and ~ 15–20%, respectively (Fig. 1h, i). However, the heights of OE plants were not significantly different from those of WT (Fig. 1j). In addition, OE plants appeared greener, especially at the mature stage, and the leaves of OE plants were thicker than those of WT. Also, Chl a, Chl b, and Car x contents of leaves increased at the seedling and mature stages of OE lines (Fig. 1k, l).

Analysis of plant phenotype. a The architecture of WT and OE (OsSPL14-overexpression) rice plants. b Leaf morphological characteristics. c Gene expression analysis of OsSPL14 in WT and OE lines. d OsSPL14 protein levels in WT and OE lines. HSP is shown as the loading control. Values below panels indicating the signal strength of OsSPL14 were quantitated using densitometry and normalized to the HSP level. The value of WT was set as one. e Lengths of flag leaves. f Widths of flag leaves. g Numbers of tillers. h Diameters of antepenultimate internodes. i Wall thicknesses of antepenultimate internodes. j Plant heights. k Chl a, Chl b, and Car x contents of leaves at the seedling stage. l Chl a, Chl b and Car x contents of flag leaves at the mature stage. Bar = 5 cm. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001)
Comparative analysis of transcriptome profiling
To investigate the impact of OsSPL14 overexpression on the rice transcriptome, we conducted an RNA sequencing experiment using OE line 1 and WT (Control). DEGs were identified using the screening standards FC ≥ 2 and FDR < 0.01. A total of 576 DEGs, containing 473 up- and 103 down-regulated genes, were identified (Fig. 2a).

Transcriptome analysis. a Volcano plot of DEGs. Larger absolute values of the abscissa indicate greater difference multiples, and larger ordinate values indicate greater significant differential expression. Each point represents a gene. Green, red, and blue indicate down-regulated, up-regulated, and non-differentiated genes, respectively. b Gene ontology classifications of DEGs. The abscissa represents the GO classification. The left side of the ordinate represents the percentage of genes and the right side represents the number of genes. c The most enriched GO terms. The ordinate represents the enriched GO terms, and the abscissa represents the q-values. The digits on the columns indicate the numbers of DEGs. Different colors represent biological processes, cellular components, and molecular functions. d Scatter diagram of KEGG enrichment. The longitudinal axis represents the pathway name, and the lateral axis represents the enrichment factor. The size of the point indicates the number of DEGs in the pathway, and the color of the point corresponds to a different q-value range
Subsequently, GO was used to classify the DEG functions. The DEGs were categorized into three major GO categories, biological process, cellular component, and molecular function, which totally contained 51 functional groups (Fig. 2b). Additionally, the determination of the most enriched GO terms was conducted by KOBAS (2.0) using FDR ≤ 0.05, which revealed that the DEGs were grouped into 20 different GO terms, including 15 terms for molecular function, three terms for biological process, and two terms for cellular component (Fig. 2c). The results implied that the DEGs annotated with GO terms were mainly related to “ion binding”, “heme binding”, “sequence-specific DNA binding”, “oxidation–reduction process”, “regulation of transcription”, and “cytoplasmic membrane-bounded”.
Moreover, to further understand the biological functions of the DEGs, we mapped the DEGs using the KEGG database and obtained 20 KEGG pathways that were the most enriched (Fig. 2d; Supplement Table S2). According to enrichment factor, Q values, and the number of enriched genes in each corresponding pathway, five KEGG pathways were selected to be annotated in detail. This analysis showed that most of the DEGs in these pathways were up-regulated (Table 1). In particular, the five DEGs in “Carotenoid biosynthesis” were all up-regulated. Among them, “BGIOSGA011548”, encoding beta-carotene hydroxylase 2, and “BGIOSGA029294”, encoding phytoene synthase, are important genes involved in carotenoid biosynthesis. Because carotenoid is the precursor of ABA, the other genes “BGIOSGA025169” and “BGIOSGA013214”, encoding 9-cis-epoxycarotenoid dioxygenase, and “BGIOSGA029635”, encoding ABA 8′-hydroxylase 3, also participate in ABA metabolism. The up-regulated DEGs in “Phenylpropanoid biosynthesis” were “BGIOSGA005999”, encoding phenylalanine ammonia-lyase (PAL), “BGIOSGA006502” and “BGIOSGA008177”, encoding trans-cinnamate 4-monooxygenase/cinnamate-4- hydroxylase (C4H), and “BGIOSGA026897”, encoding “4-coumarate-CoA ligase” (4CL), which are also key enzyme genes for lignin because lignin is a phenylpropanoid monomer polymer (Zhong and Ye 2015). The pathway showed the highest enrichment factor for “Thiamine metabolism”, in which the up-regulated DEG was “BGIOSGA025304”, encoding 1-deoxy-d-xylulose-5-phosphate synthase 2 (DXS). DXS is an important enzyme involved in terpenoid anabolism, which includes the biosynthesis of chlorophylls, tocopherols, carotenoids, ABA, and gibberellins (GAs) (Aharoni et al. 2005; Estévez et al. 2001). Additionally, the DEGs in “Plant hormone signal transduction” are related to JA, ethylene, and ABA signal transduction. The DEGs in “Starch and sucrose metabolism” are mainly related to sucrose, starch, trehalose, fructan, and pectin metabolism.
Gene expression analysis
To confirm the DEGs identified in the transcriptome analysis, several genes from the five selected KEGG pathways were analyzed by qRT-PCR. The expression levels of 15 up-regulated DEGs were higher in OsSPL14 OE line 1 than in WT (Fig. 3a), while the expression levels of five down-regulated DEGs were lower in OsSPL14 OE line 1 than in WT (Fig. 3b). Thus, the expression profiles of DEGs analyzed by qRT-PCR were consistent with the transcriptome analysis.

qRT-PCR and immunoblot analysis. a Expression patterns of 15 up-regulated genes validated by qRT-PCR. C4H (502): BGIOSGA006502; C4H (177): BGIOSGA008177. b Expression patterns of five down-regulated genes validated by qRT-PCR. c The expression profiles of TB1, DEP1, D17, D53, D14, and D3
OsSPL14 regulates the expression of TB1 and DEP1, and OsSPL14 may act with D53 to mediate SL-regulated tiller development (Lu et al. 2013; Song et al. 2017). Thus, the expression profiles of TB1, DEP1, and some genes related to SL biosynthesis or SL signal transduction were also assessed by qRT-PCR. TB1 showed no obvious change, while DEP1 was up-regulated slightly in OE line 1. In addition, compared with WT, DWARF (D) 17, which is involved in SL biosynthesis, was up-regulated in OE line 1. D14 and D3, which involved in SL signal transduction, were up-regulated in transgenic plants (Fig. 3c). Thus, the overexpression of OsSPL14 could indeed influence the expression of several genes involved in plant architecture.
Changes in ABA, JA and GA3 contents
Compared with WT, the OsSPL14 OE lines had different phenotypes. Based on the transcriptome analysis, the expression levels of several genes in carotenoid biosynthesis, ABA metabolism, terpenoid anabolism, and plant hormone signal transduction pathways are up-regulated. Thus, we wanted to determine whether the relevant plant hormone contents were altered in OE lines. Consequently, we measured ABA, JA, and GA3 contents of OE lines 1 and 2 at seedling and tillering stages. Compared with that of WT, the ABA contents of the OE lines increased at the seedling stage, by 18% and 23% in the two transgenic lines. The ABA contents of OE lines increased slightly at tillering stage (Fig. 4a). However, the JA contents showed no changes in OE lines at the seedling stage compared with WT and decreased slightly in OE line 1 at tillering stage (Fig. 4b). In contrast, the GA3 contents of the OE lines increased dramatically at the seedling stage, being approximately three times of that in WT. Although the GA3 contents of the OE lines and WT decreased significantly at tillering stage, the GA3 contents of OE lines were still 56% and 27% greater than that of WT (Fig. 4c). Thus, there are higher levels of ABA and GA3 in OE lines than that in WT.

ABA, JA, and GA3 contents in OsSPL14 OE lines and WT. a ABA contents at the seedling and tillering stages. b JA contents at the seedling and tillering stages. c GA3 contents at the seedling and tillering stages. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001)
SEM and compositional analysis of culm
One obvious characteristic of OE plants is their strong culm. Consequently, the internal organizational structure of culm in OE plants was observed using SEM. The OE plants had more small vascular bundles and sclerenchyma cells, and also had larger big vascular bundles than WT. Moreover, the degree of lignification in cortical cells was greater in OE plants (Fig. 5a).

SEM and chemical components analysis of culm in OsSPL14 OE lines and WT. a Scanning electron microscopy analysis. BVB Big vascular bundle, SVB Small vascular bundles, SC sclerenchymal cells. b Lignin content of the culm. c Cellulose content of the culm. d Silicon content of the culm. e Potassium content of the culm. (*P ≤ 0.05, **P ≤ 0.01)
In addition, the expression profiles of key genes involved in lignin biosynthesis and carbohydrate metabolism changed significantly in OE lines. Consequently, we hypothesized that the culm composition of OsSPL14 OE plant was likely to change compared with that of WT. Therefore, we measured the lignin, cellulose, Si, and K contents of the culm in OE lines 1 and 2. In contrast with that of WT, the lignin contents of the OE lines increased by 17% and 30% in the two lines, respectively (Fig. 5b). Additionally, the cellulose contents of the OE lines increased by 13% and 16% (Fig. 5c). Remarkably, the Si contents of the OE lines increased by 45% and 78%, respectively compared with that of WT (Fig. 5d). In addition, the K contents also increased by 18% and 19% in the two OE lines, respectively (Fig. 5e). Thus, overexpression of OsSPL14 in rice could cause great changes in culm characteristics and composition. The latter is closely responsible for culm mechanical strength. Thus, our results may explain the previous findings that the culm mechanical strength of NIL OsSPL14ipa1 plants with high expression levels of OsSPL14 was significantly increased (Jiao et al. 2010).
Grain quality analysis
Transgenic rice lines overexpressing OsSPL14 have large panicles, more branching, and more grains per panicle (Jiao et al. 2010; Miura et al. 2010). Here, we performed grain quality analysis of OE lines 1 and 2. The grain length, grain width, brown rice rate, head rice rate, and gelatinization temperature were not different in OsSPL14 OE lines compared with WT (Fig. 6a–e). The milled rice rate and amylose contents of OE line 2 decreased, but those of OE line1 showed no significant changes (Fig. 6f, g). However, the chalkiness ratios and chalkiness degrees of the OE lines decreased significantly compared with those of WT (Fig. 6h, i). In addition, the gel consistency levels of the OE lines improved (Fig. 6j). Thus, a few of the grain quality characteristics of the OsSPL14 OE lines were altered to a certain extent.

Grain quality analysis in OsSPL14 OE lines and WT. a Grain length. b Grain width. c Brown rice rate. d Head rice rate. e Gelatinization temperature. f Milled rice rate. g Amylose content. h Chalkiness ratio. i Chalkiness degree. j Gel consistency. (*P ≤ 0.05)
Discussion
OsSPL14, as a transcription factor, plays an important role in rice plant architecture. The typical characteristics of transgenic plants overexpressing OsSPL14 were reduced tiller number and strong culm. In addition to these characteristics, we observed that OsSPL14 OE plants have shorter growth periods. This was consistent with previous research in which the overexpression of SPL3 caused early flowering and SPL9/SPL15 controlled the juvenile-to-adult phase transition in A. thaliana (Cardon et al. 1997; Schwarz et al. 2008). SPL3 impacts flowering by directly activating the plant-specific transcription factor LEAFY, MADS domain protein FRUITFULL, and APETALA1, which control meristem identity transition (Yamaguchi et al. 2009). The SPL genes may affect flowering as downstream targets of CONSTANS and flowering time regulator at the shoot apex (Schmid et al. 2003). However, SPLs may control flowering in an endogenous flowering pathway different from the familiar flowering time regulator/bZIP transcription factor flowering model, and the miR156/SPL module may promote flowering in the absence of photoperiodic cues (Wang et al. 2009). Thus, the molecular mechanisms of the SPLs involved in controlling flowering are not fully understood, especially in rice.
OsSPL14 OE plants had short narrow flag leaves and thick green leaves in this study. In A. thaliana, most miR156-targeted SPL genes have high expression levels in the shoot apex (Cardon et al. 1997; Wu and Poethig 2006). Moreover, Wang et al. (2008) indicated that miR156 quantitatively modulates SPL expression in the leaf primordia, and SPL mediates the nonautonomous effects of existing leaf primordia on the initiation of new leaf primordia at the shoot apical meristem. Plants overexpressing SPL9 showed a strong plastochron, with a reduced leaf initiation rate. SPLs function in regulating plastochron length and leaf size, but the mechanisms, including the downstream target genes of SPLs in leaf development, are still unknown. Similarly, OsSPL14 is predominantly expressed in the shoot apex (Jiao et al. 2010; Miura et al. 2010), and OsSPL14 mRNA levels decrease in flag leaves of OsmiR156-overexpression transgenic plants (Xie et al. 2006). Thus, OsSPL14 may involve in flag leaf development, although there are limited studies on the molecular mechanisms of SPLs in regulating leaf development in rice. Additionally, we observed that the OsSPL14 OE lines were greener than WT, and the Chl a, Chl b, and Car contents were greater at the seedling and mature stages. This may be chiefly attributable to the up-regulation of genes involved in terpenoid anabolism as determined by our transcriptome analysis. Nevertheless, there is limited research on the effects of SPLs on leaf color in rice and A. thaliana.
Plant hormones play vital roles in regulating plant growth, development, and adaption to environmental changes (Santner and Estelle 2009). ABA is a phytohormone that regulates seed dormancy, seed embryo development, and stress-tolerance improvement (Nambara and Marion-Poll 2005; Matilla et al. 2015). SLs are hormones involved in suppressing lateral shoot branching, root development, response to environmental condition, and secondary growth in vascular plants (Ruyter-Spira et al. 2013; Al-Babili and Bouwmeester 2015). ABA and SLs are apocarotenoids that share carotenoid as a biosynthetic precursor (Matusova et al. 2005). Our transcriptome analysis revealed that the expression levels of the phytoene synthase and beta-carotene hydroxylase 2 genes, which are key enzyme genes in carotenoid biosynthesis, were up-regulated in OsSPL14-overexpression rice plants. In addition, the expression levels of the 9-cis-epoxycarotenoid dioxygenase gene, which participates in the rate-limiting step of ABA biosynthesis, and genes involved in SL biosynthesis and SL signal transduction also changed. Indeed, the carotenoid content of the transgenic plants was greater than that of the WT, and thus, it could provide more precursors for ABA or SL biosynthesis. Because of the common biosynthetic precursor, there is an inevitable correlation between ABA and SL levels. ABA-deficient tomato mutants exhibit greatly reduced ABA and SL contents with the downregulation of SL biosynthetic genes, suggesting a role of ABA in the regulation of SL biosynthesis (López-Ráez et al. 2010). Similarly, SL-deficient plants showed reduced ABA levels under osmotic stress (Liu et al. 2015). The biosynthetic pathways of ABA and SLs may be functionally connected. Haider et al. (2018) indicated that SLs interact with ABA during drought response in rice. SL and ABA pathways are connected through the SL biosynthetic enzyme D27, and its overexpression in rice results in increased ABA levels. In our study, OsSPL14 OE lines had higher ABA contents. Thus, we hypothesize that the higher ABA level could accompany SL to regulate plant growth and development.
In addition, GA is another important growth-promoting hormone. GA is a tetracyclic diterpenoid plant hormone that participates in diverse growth and developmental processes, including leaf differentiation, stem elongation, trichome development, seed germination, and flowering induction (Olszewski et al. 2002; Fleet and Sun 2005). One main function of GA is regulating organ elongation by affecting cell division and elongation. The GA-deficient dwarf1 (gdd1) rice mutant showed a phenotype with greatly reduced lengths of roots, stems, spikes, and seeds, which could be rescued by exogenous GA3 treatments. Furthermore, GDD1, having a transcriptional regulatory activity, affects GA accumulation by regulating ent-kaurene oxidase, which is involved in GA biosynthesis (Li et al. 2011). In Arabidopsis, SPL8, a member of the SPL gene family, has a tissue-dependent regulatory role in response to GA during plant development. Changes in SPL8 expression affects the transcription of genes involved in GA biosynthesis and signaling (Zhang et al. 2006). In this study, we found that the expression of DXS, which is involved in terpenoid anabolism, was up-regulated in the OsSPL14 OE lines, and the GA3 content increased significantly compared with in WT. The latest research indicated that GA signaling reduces the number of tillers (Liao et al. 2019), which corroborates our results. Moreover, the other main function of GA is promoting flowering. SPL15 is a key component in the integration of age- and GA-derived flowering pathways at the Arabidopsis shoot meristem (Hyun et al. 2016). Thus, we hypothesized that there was a definite correlation between early flowering in OsSPL14 OE plants and high GA levels. IPA1/OsSPL14 regulates the GA pathway and blocks GA signal transduction through SLR1 during disease resistance-related processes (Liu et al. 2019). It is probable that IPA1/OsSPL14 regulated the GA pathway through varied mechanisms in different situations and different growth stages. Thus, we inferred that the overexpression of OsSPL14 influences several plant hormones’ biosynthesis to regulate plant development. However, the mechanisms need to be further elucidated.
In vascular plants, the major component of the secondary cell wall is lignocellulose, which mainly consists of lignin, cellulose, hemicellulose, and cell wall proteins (Kumar et al. 2016). Lignin is a phenylpropanoid monomer polymer that maintains the structural integrity of the cell wall and increases the mechanical strength of the plant. The pathway of lignin monomer biosynthesis starts with the deamination of phenylalanine by PAL. Then, the product, cinnamic acid, is transformed to coumaric acid by C4H and further acted on by 4CL to produce p-coumaroyl-CoA (Boerjan et al. 2003; Zhong and Ye 2015; Kumar et al. 2016). In this study, a transcriptome analysis indicated that the expression levels of PAL, C4H, and 4CL were up-regulated in the OsSPL14 OE lines. Coincidentally, the lignin contents of the culm increased dramatically in the OsSPL14 OE lines. Cellulose is a polysaccharide, and cellulose microfibrils form the main load-bearing network (Somerville 2006). Our study suggested that the expression of some genes that participate in sugar metabolism were changed in the OsSPL14 OE lines, and the cellulose content of the culm increased, compared with WT. Guo et al. (2003) indicated that high cellulose and lignin contents within a certain stem enhanced the lodging resistance. Thus, the higher lignin and cellulose contents could enhance the mechanical strength of OsSPL14-overexpression plants. Additionally, the Si level has a close correlation with the mechanical strength of the culm. Si, which mainly exists in the epidermal layers of stems, leaf sheaths, and vascular bundles, can increase the strength and hardness of the plant cell walls (Luo et al. 2007). Increasing the Si contents of plants could improve the mechanical strengths of the basal internodes and eventually increase plant resistance to lodging (Jiang et al. 2012). Another factor, the K level, impacts the mechanical strength of the culm. Yang et al. (2004) indicated that the thickness of the basal stem and the anti-fracture characteristic were correlated with Si and K contents. Here, the Si and K contents increased in the OsSPL14 OE lines. This further illustrated that as the mechanical strength of OsSPL14-overexpression plants increased, so did the lodging resistance. Thus, OsSPL14 is a pleiotropic regulator that participates in several metabolic pathways and plays important roles in the growth and development of plants. Certainly, the regulatory network of OsSPL14 is worth further studying in rice.
References
Aharoni A, Jongsma MA, Bouwmeester HJ (2005) Volatile science? Metabolic engineering of terpenoids in plants. Trends Plant Sci 10(12):594–602
Al-Babili S, Bouwmeester HJ (2015) Strigolactones, a novel carotenoid-derived plant hormone. Annu Rev Plant Biol 66(1):161–186
Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Annu Rev Plant Biol 54(1):519–546
Cardon GH, Höhmann S, Nettesheim K, Saedler H, Huijser P (1997) Functional analysis of the Arabidopsis thaliana SBP-box gene SPL3: a novel gene involved in the floral transition. Plant J 12(2):367–377
Dai WM, Zhang KQ, Duan BW, Sun CX, Zheng KL, Cai R, Zhuang JY (2005) Rapid determination of silicon content in rice (Oryza sativa). Chin J Rice Sci 19(5):460–462
Dai ZY, Wang J, Yang XF, Lu H, Miao XX, Shi ZY (2018) Modulation of plant architecture by the miR156f-OsSPL7-OsGH3.8 pathway in rice. J Exp Bot 69(21):5117–5130
Duan E, Wang Y, Li X, Lin Q, Zhang T, Wang Y, Zhou C, Zhang H, Jiang L, Wang J, Lei C, Zhang X, Guo X, Wang H, Wan J (2019) OsSHI1 Regulates plant architecture through modulating the transcriptional activity of IPA1 in Rice. Plant Cell 31(5):1026–1042
Estévez JM, Cantero A, Reindl A, Reichler S, León P (2001) 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem 276(25):22901–22909
Fleet CM, Sun TP (2005) A DELLAcate balance: the role of gibberellin in plant morphogenesis. Curr Opin Plant Biol 8(1):77–85
Gandikota M, Birkenbihl RP, Höhmann S, Cardon GH, Saedler H, Huijser P (2007) The miRNA156/157 recognition element in the 3' UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J 49(4):683–693
Guo YH, Zhu SG, Zhang LB, Dou H (2003) Influence on the material characteristics of rice culms in different cultivation conditions. J Shenyang Agric Univ 34(1):4–7
Haider I, Andreo-Jimenez B, Bruno M, Bimbo A, Floková K, Abuauf H, Ntui VO, Guo X, Charnikhova T, Al-Babili S, Bouwmeester HJ, Ruyter-Spira C (2018) The interaction of strigolactones with abscisic acid during the drought response in rice. J Exp Bot 69(9):2403–2414
Hyun Y, Richter R, Vincent C, Martinez-Gallegos R, Porri A, Coupland G (2016) Multi-layered regulation of SPL15 and cooperation with SOC1 integrate endogenous flowering pathways at the Arabidopsis shoot meristem. Dev Cell 37(3):254–266
GB/T 13885–2003/ISO 6869:2000, ISO 6869:2000 (2000) Animal feeding stuffs-determination of the contents of calcium, copper, iron, magnesium, manganese, potassium, sodium and zinc-method using atomic absorption spectrometry. Iso International Standard
Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23(12):4185–4207
Jiang YZ, Shen XH, Cao LY (2012) Research progress of stem traits in rice. China Rice 18(2):1–7
Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42(6):541–544
Kim SR, Ramos JM, Hizon RJM, Ashikari M, Virk PS, Torres EA, Nissila E, Jena KK (2018) Introgression of a functional epigenetic OsSPL14WFP allele into elite indica rice genomes greatly improved panicle traits and grain yield. Sci Rep-UK 8(1):3833
Klein J, Saedler H, Huijser P (1996) A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol Gen Genet Mgg 250(1):7–16
Kumar M, Campbell L, Turner S (2016) Secondary cell walls: biosynthesis and manipulation. J Exp Bot 67(2):515–531
Lee J, Park JJ, Kim SL, Yim J, An G (2007) Mutations in the rice liguleless gene result in a complete loss of the auricle, ligule, and laminar joint. Plant Mol Biol 65(4):487–499
Li J, Jiang J, Qian Q, Xu Y, Zhang C, Xiao J, Du C, Luo W, Zou G, Chen M, Huang Y, Feng Y, Cheng Z, Yuan M, Chong K (2011) Mutation of rice BC12/GDD1, which encodes a kinesin-like protein that binds to a GA biosynthesis gene promoter, leads to dwarfism with impaired cell elongation. Plant Cell 23(2):628–640
Li M, Li X, Zhou Z, Wu P, Fang M, Pan X, Lin Q, Luo W, Wu G, Li H (2016) Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front in Plant Sci 7(12217):377
Liao Z, Yu H, Duan J, Yuan K, Yu C, Meng X, Kou L, Chen M, Jing Y, Liu G, Smith SM, Li J (2019) SLR1 inhibits MOC1 degradation to coordinate tiller number and plant height in rice. Nat Commun 10(1):2738
Liu J, He H, Vitali M, Visentin I, Charnikhova T, Haider I, Schubert A, Ruyter-Spira C, Bouwmeester HJ, Lovisolo C (2015) Osmotic stress represses strigolactone biosynthesis inLotus japonicus roots: exploring the interaction between strigolactones and ABA under abiotic stress. Planta 241(6):1435–1451
Liu M, Shi Z, Zhang X, Wang M, Zhang L, Zheng K, Liu J, Hu X, Di C, Qian Q, He Z, Yang DL (2019) Inducible overexpression of Ideal Plant Architecture 1 improves both yield and disease resistance in rice. Nat Plants 5(4):389–400
López-Ráez JA, Kohlen W, Charnikhova T, Mulder P, Undas AK, Sergeant MJ, Verstappen F, Bugg TDH, Thompson AJ, Ruyter-Spira C, Bouwmeester H (2010) Does abscisic acid affect strigolactone biosynthesis? New Phytol 187(2):343–354
Lu Z, Yu H, Xiong G, Wang J, Jiao Y, Liu G, Jing Y, Meng X, Hu X, Qian Q, Fu X, Wang Y, Li J (2013) Genome-wide binding analysis of the transcription activator ideal plant architecture1 reveals a complex network regulating rice plant architecture. Plant Cell 25(10):3743–3759
Luo MC, Tian CT, Li XJ, Lian JX (2007) Relationship between morpho-anatomical traits together with chemical components and lodging resistance of stem in rice (Oryza sativa L). Acta Bot Boreale-Occidentalia Sin 27(11):2346–2353
Matilla AJ, Carrillo-Barral N, del Carmen Rodriguez-Gacio M (2015) An update on the role of NCED and CYP707A ABA metabolism genes in seed dormancy induction and the response to after ripening and nitrate. J Plant Growth Regul 34(2):274–293
Matusova R, Rani K, Verstappen FW, Franssen MC, Beale MH, Bouwmeester HJ (2005) The strigolactone germination stimulants of the plant-parasitic Striga and Orobanche spp. are derived from the carotenoid pathway. Plant Physiol 139(2):920–934
Miura K, Ikeda M, Matsubara A, Song XJ, Ito M, Asano K, Matsuoka M, Kitano H, Ashikari M (2010) OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat Genet 42(6):545–549
Nambara E, Marion-Poll A (2005) Abscisic acid biosynthesis and catabolism. Annu Rev Plant Biol 56(1):165–185
Olszewski N, Sun TP, Gubler F (2002) Gibberellin signaling: biosynthesis, catabolism, and response pathways. Plant Cell 14(Suppl):S61–80
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110(4):513–520
Ruyter-Spira C, Al-Babili S, van der Krol S, Bouwmeester H (2013) The biology of strigolactones. Trends Plant Sci 18(2):72–83
Santner A, Estelle M (2009) Recent advances and emerging trends in plant hormone signaling. Nature 459(7250):1071–1078
Schmid M, Uhlenhaut NH, Godard F, Demar M, Bressan R, Weigel D, Lohmann JU (2003) Dissection of floral induction pathways using global expression analysis. Development 130(24):6001–6012
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8(4):517–527
Schwarz S, Grande AV, Bujdoso N, Saedler H, Huijser P (2008) The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol Biol 67(1–2):183–195
Shao YL, Zhou HZ, Wu YR, Zhang H, Lin J, Jiang XY, He QJ, Zhu JS, Li Y, Yu H, Mao CZ (2019) OsSPL3, an SBP-domain protein, regulates crown root development in rice. Plant Cell 31(6):1257–1275
Somerville C (2006) Cellulose synthesis in higher plants. Annu Rev Cell Dev Bi 22(1):53–78
Song X, Lu Z, Yu H, Shao G, Xiong J, Meng X, Jing Y, Liu G, Xiong G, Duan J, Yao XF, Liu CM, Li H, Wang Y, Li J (2017) IPA1 functions as a downstream transcription factor repressed by D53 in strigolactone signaling in rice. Cell Res 27(9):1128–1141
Sun H, Tao J, Liu S, Huang S, Chen S, Xie X, Yoneyama K, Zhang Y, Xu G (2014) Strigolactones are involved in phosphate- and nitrate-deficiency-induced root development and auxin transport in rice. J Exp Bot 65(22):6735–6746
Tong GL, Lu Q, Wang J, Zhou CH (2005) Spectrophotometeric determination of silicon in rice straw and black liquor after rice straw alkaline pulping by silicomolybdate blue. China Pulp Paper Ind 26(8):64–66
Unte US, Sorensen AM, Pesaresi P, Gandikota M, Leister D, Saedler H, Huijser P (2003) SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell 15(4):1009–1019
Wang JW, Schwab R, Czech B, Mica E, Weigel D (2008) Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 20(5):1231–1243
Wang JW, Czech B, Weigel D (2009) miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138(4):738–749
Wang J, Yu H, Xiong G, Lu Z, Jiao Y, Meng X, Liu G, Chen X, Wang Y, Li J (2017a) Tissue-specific ubiquitination by IPA1 INTERACTING PROTEIN1 modulates IPA1 protein levels to regulate plant architecture in rice. Plant Cell 29(4):697–707
Wang S, Wu K, Qian Q, Liu Q, Li Q, Pan Y, Ye Y, Liu X, Wang J, Zhang J, Li S, Wu Y, Fu X (2017b) Non-canonical regulation of SPL transcription factors by a human OTUB1-like deubiquitinase defines a new plant type rice associated with higher grain yield. Cell Res 27(9):1142–1156
Wang J, Zhou L, Shi H, Chern M, Yu H, Yi H, He M, Yin J, Zhu X, Li Y, Li W, Liu J, Wang J, Chen XQ, Qing H, Wang Y, Liu G, Wang W, Li P, Wu X, Zhu L, Zhou JM, Ronald PC, Li S, Li J, Chen X (2018) A single transcription factor promotes both yield and immunity in rice. Science 361(6406):1026–1028
Wu G, Poethig RS (2006) Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 133(18):3539–3547
Xie K, Wu C, Xiong L (2006) Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol 142(1):280–293
Yamaguchi A, Wu MF, Yang L, Wu G, Poethig RS, Wagner D (2009) The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev Cell 17(2):268–278
Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Nunokawa E, Ishizuka Y, Terada T, Shirouzu M, Osanai T, Tanaka A, Seki M, Shinozaki K, Yokoyama S (2004) A novel zinc-binding motif revealed by solution structures of DNA-binding domains of Arabidopsis SBP family transcription factors. J Mol Biol 337(1):49–63
Yang CM, Yang LZ, Yan TM, Ou YZ (2004) Effects of nutrient and water regimes on lodging resistance of rice. Chin J Appl Ecol 15(4):646–650
Yue E, Li C, Li Y, Liu Z, Xu JH (2017) MiR529a modulates panicle architecture through regulating SQUAMOSA PROMOTER BINDING-LIKE genes in rice (Oryza sativa). Plant Mol Biol 94(4–5):469–480
Zhang Y, Schwarz S, Saedler H, Huijser P (2006) SPL8, a local regulator in a subset of gibberellin-mediated developmental processes in Arabidopsis. Plant Mol Biol 63(3):429–439
Zhang L, Yu H, Ma B, Liu GF, Wang JJ, Wang JM, Gao RC, Li JJ, Liu JY, Xu J, Zhang YY, Li Q, Huang XH, Xu J, Li JL, Qian Q, Han B, He ZH, Li JY (2017) A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nat Commun 8:14789
Zhong RQ, Ye ZH (2015) Secondary cell walls: biosynthesis, patterned deposition and transcriptional regulation. Plant Cell Physiol 56(2):195–214
Acknowledgements
The plasmid pCAMBIA1300-OsSPL14 was provided by Professor Jiayang Li, Institute of Genetics and Developmental Biology, Chinese Academy of Science, China. This work was supported by The Key Program of National Transgenic Research of China (Grant No. 2016ZX08001-004) and The Special Foundation of Non-Profit Research Institutes of Fujian Province (Grant No. 2016R1020-8).
Author information
Authors and Affiliations
Contributions
LL and JFZ conceived and designed the research. LL, HZ, WH, LYW, XPX, YMZ, YDW and YSZ performed experiments. YLL and LYP completed the field work. HBX and QHC analyzed the data. HAX guided and supported the research. LL wrote the manuscript. JFZ revised the paper. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lian, L., Xu, H., Zhang, H. et al. Overexpression of OsSPL14 results in transcriptome and physiology changes in indica rice ‘MH86’. Plant Growth Regul 90, 265–278 (2020). https://doi.org/10.1007/s10725-019-00569-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10725-019-00569-0