
Abstract
Metnase is a fusion gene comprising a SET histone methyl transferase domain and a transposase domain derived from the Mariner transposase. This fusion gene appeared first in anthropoid primates. Because of its biochemical activities, both histone (protein) methylase and endonuclease, we termed the protein Metnase (also called SETMAR). Metnase methylates histone H3 lysine 36 (H3K36), improves the integration of foreign DNA, and enhances DNA double-strand break (DSB) repair by the non-homologous end joining (NHEJ) pathway, potentially dependent on its interaction with DNA Ligase IV. Metnase interacts with PCNA and enhances replication fork restart after stalling. Metnase also interacts with and stimulates TopoIIα-dependent chromosome decatenation and regulates cellular sensitivity to topoisomerase inhibitors used as cancer chemotherapeutics. Metnase has DNA nicking and endonuclease activity that linearizes but does not degrade supercoiled plasmids. Metnase has many but not all of the properties of a transposase, including Terminal Inverted Repeat (TIR) sequence-specific DNA binding, DNA looping, paired end complex formation, and cleavage of the 5′ end of a TIR, but it cannot efficiently complete transposition reactions. Interestingly, Metnase suppresses chromosomal translocations. It has been hypothesized that transposase activity would be deleterious in primates because unregulated DNA movement would predispose to malignancy. Metnase may have been selected for in primates because of its DNA repair and translocation suppression activities. Thus, its transposase activities may have been subverted to prevent deleterious DNA movement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transposons are ancient mobile DNA elements that encode the enzymatic machinery for their mobility, and are found in genomes of species from bacteria to mammals (Miskey et al. 2005). These mobile elements move within genomes by two major mechanisms: (1) an excision and ligation strategy utilized by DNA transposons, and (2) by forming RNA intermediates in the case of retrotransposons. DNA transposon sequences may comprise up to 3% of the human genome, whereas retrotransposon sequences may comprise ~41% of the human genome (Lander et al. 2001). The ratio of these two large classes differs significantly among species. The Tc1/Mariner family is one of the most widely studied DNA transposons. Its members are present as defective remnants in vertebrate species. There are three active Mariner transposon elements observed in insects, while there are two types of Mariner remnants in primates, designated Hsmar1 and Hsmar2. Mariner transposons entered primate genomes approximately 50 million years ago, and remained mobile until 37 million years ago. They are between 1,300 and 2,400 bp in length and comprise a central region encoding the transposase activity, and two flanking Terminal Inverted Repeats (TIR) that mediate the binding of transposase protein to the transposon sequences, which results in the excision and ligation-mediated genome mobility of active transposons (Plasterk et al. 1999).
The Tc1/Mariner family usually integrates into TA target dinucleotides in TIR sequences in the host genome (Plasterk et al. 1999). Phylogenetic relationships among these transposons are inconsistent with those of their hosts, so that very similar Tc1/Mariner transposons can be found in very different organisms such as insects and primates. This latter observation has been explained by the horizontal spread of these transposable elements between species, which took place millions of years ago in vertebrates. The modes of transmission between species are unknown, but potential vectors would be viruses and intracellular parasites (Miskey et al. 2005), although DNA transposons are not directly infectious. Following initial transmission, the transposons would have to colonize germ lines to persist across generations within a species. During this initial period, massive amplification of these elements takes place. Subsequent mutations accumulate, resulting in defective copies in a process called vertical inactivation.
There is no known transposase activity documented in primates, with the possible exception of the transposition-like reaction mediated by the two RAG recombinases during immunoglobulin and T-cell receptor formation. The human genome project has identified 47–50 genes that could have originated from transposons, 43 of them from DNA transposons (Lander et al. 2001). It was hypothesized that these transposon genes have been inactivated, with the exception of the RAG proteins, because unregulated DNA mobility would be deleterious to primates, producing either apoptosis or malignancy (Beck et al. 2008; Lander et al. 2001).
The transposase family of enzymes contains a catalytic domain termed the DDE or DDD motif, that includes three conserved amino acids shared by transposases, RAG proteins, HIV integrase, RNAse H, and the RNA-induced silencing complex (RISC) (Dreyfus 2006). Since transposons mediate chromosomal translocations in certain organisms such as maize (Zhang et al. 2009), we theorized that a transposase activity might play a role in chromosomal translocations seen in neoplasia in primates. We focused on an EST clone (first noted by Robertson and Zumpano 1997) that encoded a putative open reading frame that was designated SETMAR by the National Center for Biotechnology Information (NCBI). Most transposase sequences in the human genome are pseudogenes, and not translated; so we isolated and characterized the protein encoded by this sequence, to prove that it was indeed translated into a functional protein. We then characterized its biochemical activities, and termed it Metnase (Lee et al. 2005). This gene represents a fusion between a SET histone methylase domain and a transposase sequence element from Hsmar1 of the human Mariner transposons. The various functions of Metnase are documented in Table 1.
Metnase structure and evolutionary history
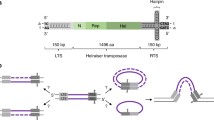
Metnase has three exons that extend over 13.8 Kb located at 3p26 (Lee et al. 2005). There are multiple types of human malignancies with cytogenetic abnormalities in this region, notably non-Hodgkin’s lymphoma, acute leukemias, hereditary prostate cancer, myeloma, and myelodysplastic syndromes. Assessing Metnase gene sequence and function in these malignancies is an important area of future study. Two exons form the SET, pre-SET and post-SET domains (Fig. 1). The third exon is the Mariner transposase sequence, which is not flanked by a TIR sequence. The amino acid sequence of the transposase is most similar to Planarian Mariner-9, with which it has 50% identity. The SET domain is most related to SuvHar91 histone methyl transferase, with 36% homology. The crystal structure of Metnase has been deposited in the structure database (histone methylase domain 3B05, transposase domain 3F2k) although not published, and it appears that Metnase exists as a dimer. We have confirmed this dimerization biochemically (unpublished).

Metnase gene structure: Exons 1 and 2 encode the pre-SET, SET and the post-SET domains. Exon 3 encodes the transposase domain, which includes an HTF motif that mediates TIR binding. The catalytic activity of transposases lies in the DDD/E-like motif, which in Metnase is DDN, which may account for the lack of certain transposase activities. Metnase is phosphorylated at S495 by Chk1 after DNA damage (unpublished data)
One report detailed the evolution of this fusion gene (Cordaux et al. 2006). Metnase exists only in anthropoid primates (Humans, apes, and old and new world monkeys). It is not found in prosimians or lower mammals. It appears to have emerged 40–58 million years ago. Detailed genetic analysis clarified the mechanism of exon capture of the coding region of Hsmar1 transposase and the creation of a fusion gene with the two SET exons (Cordaux et al. 2006). It is interesting to note that this same period witnessed the significant transposon invasion and expansion in primates (Lander et al. 2001; Miskey et al. 2005). By comparing the rate of non-synonymous (K A) and synonymous (K S) nucleotide substitutions per site in the transposase region of each anthropoid lineage, it was shown that the K A/K S ratios among different primate lineages were not significantly different. This suggested that the Mariner region has evolved consistently under purifying selection in all anthropoid lineages. By evaluating different regions within the transposase DNA, it was found that the 5′ half of the exon 3 transposase sequence (the TIR binding region) displayed a strong signal of purifying selection (K A/K S = 0.1, significantly <1), whereas the 3′ half (the catalytic region) displayed a signal of nearly neutral evolution (K A/K S = 0.7), suggesting that the Mariner portion of Metnase may have been recruited for a function located in the N-terminal DNA binding region of the transposase (Cordaux et al. 2006). However, as we describe below, Metnase retains some catalytic activity of its transposase domain, at least in vitro in various experimental settings.
Histone methylase activity of Metnase
Metnase stimulates the dimethylation of histone H3 at lysine 36 (H3K36me2), and to a lesser extent H3 on lysine 4 (H3K4me2) (Lee et al. 2005). Histone methylation has emerged as a major regulator of chromatin functions, such as transcription, DNA repair, DNA replication, imprinting, and X-chromosome inactivation (Kouzarides 2007). H3K4 and H3K36 methylation have been associated with chromatin that is accessible to transcription regulators and DNA repair proteins (Bernstein et al. 2005). The current evidence indicates that histone methylation creates docking sites for other proteins, recruiting them for transcription or repair. For example, H3K36 methylation attracts the histone deacetylase Rpd3S, which compacts chromatin in the middle of transcribed genes, and inhibits spurious initiation of transcription during the elongation phase (Carrozza et al. 2005). While H3K4 methylation has been noted largely in the promoter areas of genes, H3K36 methylation peaks in the middle and towards the 3` end of transcribed genes (Bernstein et al. 2007). While the consequences of H3K36 methylation are clear as far as suppression of spurious transcription initiation, its impact on activation of gene transcription is not as clear. The methyltransferase Setd2 (also termed Set2) mediates trimethylation of H3K36, and it binds the phosphorylated tail of RNA polymerase II, which implicates H3K36 in transcription (Li et al. 2002). Setd2 mediates H3k36 trimethylation in mammalian cells, but not di- or mono-methylation (Edmunds et al. 2008). These data raise the possibility that Metnase or other H3K36 dimethyltransferases (G9a, Smyd2) may be needed to create dimethylated H3K36 before Setd2 acts. In Drosophila, dimethylation of H3K36 peaks adjacent to promoters and requires distinct methyltransferases than those that mediate trimethylation of H3K36, which accumulates toward the 3′ end of genes (Bell et al. 2007).
Metnase may play a role in other histone modifications besides H3K36me2. Metnase was shown to interact with Pso4 (also termed Prp19) in a co-immunoprecipitation assay (Beck et al. 2008). The interaction of Metnase with Pso4 is essential for the localization of Metnase to sites of DNA DSBs, and is therefore important for Metnase DNA repair activity. However, Pso4 is a component of a complex instrumental in RNA splicing, which takes place in the vicinity of the transcriptional machinery on nascent RNA. This association of Metnase with Pso4 suggests a potential role in RNA transcription and splicing through methylation of H3K36 or other protein targets. As discussed below, the Metnase transposase domain has potent DNA looping activity in vitro (Liu et al. 2007). It would be interesting to determine if this activity was involved in RNA looping during splicing. The evolution of prosimians to anthropoid primates may have been associated with a selective pressure for more accurate and efficient transcription and splicing, in order to provide novel protein functions with a similar number of gene sequences (Kriventseva et al. 2003). The activity of Metnase on H3K4 is less prominent than H3K36, at least in vitro. It is unknown if other factors enhance this activity in vivo (Fig. 2).

Potential Metnase binding sites in the human genome. There are about 200 copies of the defective Mariner ancestor Hsmar1, about 2,500 copies of MITES, which are similar to Tc1/Mariner transposons but lack central elements. There are also about 4,500 solo TIR sequences
Residual transposase functions of Metnase
Metnase binds to a specific 19 bp sequence within the consensus Hsmar1 TIR (Kouzarides 2007). Similar to other Mariner transposases, the Metnase Helix-Turn-Helix (HTH) motif accounts for this binding; specifically the R432 residue within the HTH region is essential for this binding (Roman et al. 2007). In human genomes there are a large number of miniature inverted-repeat transposable elements (MITES). If the solo TIRs are added to the number of MITES, there are approximately 7,000 potential Metnase binding sites within the human genome. But as described above (Fig. 1), Metnase binds to Pso4, which can bind indiscriminately to double-stranded DNA. Thus, its interaction with Pso4 could theoretically provide non-specific access of Metnase to DNA (Beck et al. 2008). We mentioned earlier that synonymous versus non-synonymous mutation analysis of the Metnase transposase sequence suggested a selective pressure towards preserving the DNA binding region rather than the catalytic domain, consistent with its ability to enhance DNA repair. However, Metnase still retains many characteristics of ancestral Mariner transposases. These include TIR DNA binding, cleavage of 5′ end of the transposon in the presence of Mn2+, DNA looping, assembly of a paired end complex, and integration at TA sites (Liu et al. 2007; Miskey et al. 2007). Metnase is efficient in 5′ nicking of Hsmar1 transposons in vitro. However, Metnase displayed a severe defect in cleavage of 3′ ends, which limits its ability to mobilize transposons in vivo, and makes it unlikely that Metnase could transpose DNA elements in primates (Liu et al. 2007). This defective 3′ nicking phenotype was also simultaneously demonstrated by another group, who suggested that Metnase generates 5′ nicks rather than complete DSBs, which they found could be induced by reconstructed functional Hsmar1 transposase (Miskey et al. 2007) (Fig. 3).

Role of Metnase in NHEJ. Metnase binds DNA DSBs in the presence of Pso4, and then interacts with DNA Ligase IV, which catalyzes the final ligation step. This figure does not include the potential ways by which Metnase could enhance NHEJ repair by modifying chromatin structure through its H3K36 histone methylase activity. As noted in the text, SET domain mutations interfere with the ability of Metnase to enhance NHEJ, implying that histone methylation is indeed important for its activity in NHEJ repair. The crystal structure of Metnase has been deposited in the structure database (3B05, 3F2k) although not published, and it appears that Metnase exists as a dimer
The Metnase transposase catalytic domain does not preserve the conserved DDD (Asp in the carboxy terminal position) or DDE (Glu) triplet motifs of active transposases; rather it has a DDN (Asn) configuration. Isolated exon 3 protein of Metnase, encoding the transposase domain, resulted in extremely weak in vitro transposition of a reconstructed Hsmar1 transposon (Liu et al. 2007). Restoration of the DDD motif in the exon 3 protein slightly increased the frequency of transposition (Liu et al. 2007). While Metnase does not have full endonuclease activity for linear DNA, we found that it does indeed have endonuclease activity for DNA with tertiary structure, including supercoiled DNA. Metnase converts supercoiled plasmid DNA to relaxed circles, and to a lesser extent to linear forms, but it does not further degrade linear DNA (Roman et al. 2007). The D483 residue within DDN motif in Metnase is essential for these endonuclease and nicking activities, but TIR sequences are not required and Metnase appears to cleave supercoiled DNA without any sequence specificity (Miskey et al. 2007; Roman et al. 2007). Thus, with linear DNA, Metnase has 5′ endonuclease activity that is specific for TIR sequences, but with supercoiled DNA it has non-specific nicking (endonucleolytic) activity.
Metnase in foreign DNA integration
Since Metnase encodes a transposase domain (albeit a defective one) that has similarity to the catalytic domains of retroviral integrases, we postulated that Metnase might enhance viral DNA integration into host genomic DNA. We found that Metnase enhances Molony Leukemia virus DNA integration by fourfold (Lee et al. 2005). It also promotes plasmid DNA integration by 2.6 to 6 fold, whether the integrating plasmid expresses Metnase (cis integration) or on a separate plasmid (trans integration) (Lee et al. 2005). Metnase is also capable of further enhancing foreign DNA integration into DSBs induced by I-SceI nuclease (unpublished results). Conversely, reducing Metnase levels by siRNA decreased plasmid and viral DNA integration (Lee et al. 2005). This led us to examine whether Metnase levels influenced genomic integration of intact HIV infecting human cells. We found that Metnase over-expression was accompanied by an increase in the number of copies of integrated HIV cDNA, whereas reducing the Metnase level decreased the amount of HIV cDNA copies integrated in the target human cell genomes (Williamson et al. 2008a). Metnase had no effect on the amount of either total cellular viral RNA, cDNA, or double-LTR circles after infection, indicating that Metnase increases cDNA integration into the host genome, without affecting other infection steps. Thus, Metnase plays a role in the integration of foreign DNA into host human cell genomes, and it may therefore be a valuable target for therapeutic suppression of HIV infection. HIV preferentially targets certain regions of the human genome and Metnase does not influence this preference. It is not yet known whether Metnase promotes random integration of plasmid DNA and it is possible that Metnase preferentially integrates DNA into the ~7,000 Mariner remnants in the human genome. Non-targeted DNA integration is a major obstacle to efficient, homologous recombination-directed gene targeting in human cells. Knocking down Metnase or chemically inhibiting its integration activity holds promise for enhancing gene targeting that may direct the development of safer and more effective gene therapy strategies.
Metnase in DNA repair
We hypothesized that Metnase, given its histone modifying activity, and its DNA binding and cleavage activity, could play a role in DNA repair. We focused on DSBs since they are required for chromosomal translocations, and since DSBs enhance exogenous DNA integration, a function to which Metnase contributes (Lee et al. 2005). DSBs are repaired by two major pathways: homologous recombination (HR) and non-homologous end joining (NHEJ). Metnase over-expression had no impact on DSB repair by HR, but it significantly enhanced NHEJ (Lee et al. 2005). Metnase knockdown by siRNA reduced imprecise NHEJ by 12-fold and precise NHEJ by 20-fold. Mutations in essential residues within the SET or the transposase domains abrogated the NHEJ enhancing activity of Metnase. Interestingly, a single substitution (D490S) in the transposase domain had a dominant negative effect, repressing NHEJ below levels seen with endogenous Metnase (Fig. 3). Metnase overexpression also enhances cell survival after exposure to ionizing radiation, which generates DSBs (Lee et al. 2005). We therefore addressed whether Metnase interacts with known components of the NHEJ pathway. We found that Metnase co-immunoprecipitates with the essential NHEJ component DNA Ligase IV (Hromas et al. 2008), which mediates the final re-ligation step in NHEJ repair. As mentioned above, Metnase interacts with Pso4 (Beck et al. 2008). Pso4 is a multifunctional protein implicated in RNA splicing and DNA repair (Mahajan and Mitchell 2003) among other processes. Pso4 binds double-stranded DNA and forms a stable complex with Metnase on both TIR and non-TIR DNA, and this interaction is required for the activity of Metnase activity in NHEJ repair. Both proteins co-localize with Nbs1 foci at DSB regions, and knocking down Pso4 with siRNA ablated Metnase localization at DSB foci (Beck et al. 2008). Of note, Pso4 is involved in repair of other types of DNA lesions, such as inter-strand cross-links (Zhang et al. 2005). Pso4 is also present in a complex with the XAB2 protein which is important in transcription-coupled repair of DNA (Kuraoka et al. 2008). This last interaction, in addition to Pso4 role in RNA splicing, and the role of Metnase in H3k36 methylation (which correlates with transcription elongation), suggests potential functions for Metnase and Pso4 in transcription-coupled nucleotide excision repair.
Metnase may have roles in other DNA repair processes. Endonucleases in general contribute to NHEJ repair by processing the free DNA ends, and to nucleotide excision repair by excising single-strand DNA flanking bulky DNA lesions. Pso4 has been implicated in repair of DNA cross-links, and Metnase nicking activity could contribute to processing of these types of lesions. During NHEJ, Metnase over-expression decreases the length of the deletions seen at the repair site (Hromas et al. 2008), perhaps reflecting reduced end-processing due to accelerated repair. It is unclear whether Metnase enhancement of DNA repair is mediated primarily through its histone methylation or DNA endonuclease activities, but as mentioned above mutations in either of these two domains decrease NHEJ repair. Of note, histone methylation in other settings has been shown to create docking sites for DNA repair proteins, such as H4K20 methylation that attracts 53BP1 (Botuyan et al. 2006). We have unpublished evidence that Metnase increases the presence of H3K36Me2 at the DSB using chromatin immunoprecipitation, and that this methylation of H3K36 assists in recruiting NHEJ components, such as the MRN complex and DNA Ligase IV, to the DSB. This may be ultimately the major role of Metnase in NHEJ repair.
Metnase in decatenation and chemotherapy resistance
Given its endonucleolytic activity on supercoiled DNA, we postulated a role for Metnase in chromosomal decatenation. DNA replication results in intertwined sister chromatids that must be untangled, or decatenated, to ensure proper chromatid segregation in mitosis, and to prevent catastrophic chromatid breaks during anaphase (Ashworth 2005). TopoIIα is the principal decatenating enzyme. It functions by creating transient DSBs through which it passes double-stranded DNA (Roca 2009). We demonstrated that Metnase interacts with TopoIIα by co-immunoprecipitation, that these proteins colocalize on condensed chromosomes, and that Metnase enhances TopoIIα decatenation activity (Williamson et al. 2008b). Nuclear extracts containing Metnase decatenated highly interlocked (kinetoplast) DNA to relaxed circular forms more rapidly than those lacking Metnase, and neutralizing anti-sera against Metnase reversed that effect. Overexpression of Metnase was found to promote progression through the metaphase decatenation checkpoint induced by TopoIIα inhibitors (Williamson et al. 2008b; Wray et al. 2009a, b). We have preliminary data indicating that Metnase promotes survival after treatment with agents that cause DNA replication stress by enhancing restart of stalled and/or collapsed replication forks. These roles in the replication stress response may also depend on its interaction with TopoIIα, which may be important for relaxing positive supercoils in front of progressing replication forks (McClendon et al. 2005).
We recently demonstrated that Metnase is expressed at high levels in several acute myeloid leukemia (AML) and breast cancer cell lines (Wray et al. 2009a, b). In the AML cells, Metnase was expressed at much higher levels than in CD34+ hematopoietic progenitors, the normal cell counterpart to AML cells (Wray et al. 2009a). In both the breast cancer and AML cells, we showed that Metnase interacts with TopoIIα. Also, we found that knocking down Metnase significantly increased metaphase decatenation checkpoint arrest induced by the TopoIIα inhibitor ICRF-193 in both the breast cancer and AML cells (Wray et al. 2009a, b). Importantly, knocking down Metnase markedly sensitized breast cancer cells to the TopoIIα inhibitor adriamycin, which is widely used in the treatment of breast cancer. In like manner, knocking down Metnase markedly sensitized AML cells to the widely used leukemia drug etoposide. Also, adding Metnase to TopoIIα blocked the inhibitory effects of both adriamycin and etoposide on TopoIIα-dependent decatenation in vitro (Wray et al. 2009a, b). These results suggest Metnase as a critical target for small molecule inhibition to enhance the effects of cancer chemotherapy, at least those involving with TopoIIα inhibitors.
Metnase in replication fork restart
Using BRDU incorporation and flow cytometry, we found that Metnase promotes cell proliferation, but it does not alter cell cycle phase distributions, or the speed of replication fork progression. However, Metnase knock-down greatly sensitized cells to replication stress induced by hydroxyurea, UV light, and the topoisomerase I inhibitor camptothecin. Such knock-downs conferred a marked defect in restart of stalled replication forks. Metnase also promotes resolution of phosphorylated H2Ax foci, a marker of DNA double-strand breaks at collapsed forks. Metnase was also found to co-immunoprecipitates with PCNA, and RAD9, a member of the PCNA-like RAD9-RAD1-HUS1 (9-1-1) complex that has key roles in the intra-S checkpoint, and the restart of stalled forks. Using DNA fiber analysis, we found that altering Metnase levels did not alter the rate of replication fork progression, but did correlate with the rate at which forks could restart after stalling in the presence of hydroxyurea. Reducing Metnase levels decreased significantly the ability of stalled replication forks to restart, as determined with DNA fiber analysis (DeHaro et al. 2010). Metnase also promotes TopoIIα-mediated relaxation of positively supercoiled DNA, which would decrease torsional stress in front of replication forks. Together these results establish Metnase as a key factor that promotes restart of both stalled and collapsed replication forks. This could be due to enhanced TopoIIα activity in front of the stalled fork, promoting PCNA stabilization of the replisome at the stalled fork, and/or promoting 9-1-1 complex activity at the stalled fork. Interestingly, in unpublished experiments, we have recently found that Metnase is phosphorylated at S495 by Chk1 after ionizing radiation or hydroxyurea, consistent with its role in replication fork restart. Of course, the ability of Metnase to promote restart of stalled replications forks could be due to its intrinsic histone methylase or endonuclease activities as well.
Conclusion
Metnase is a unique and intriguing transposase-related protein for many reasons: (1) it is present only in primates, (2) it fuses histone methylase activity with DNA structure-specific endonuclease activity, (3) it enhances DNA repair by NHEJ, (4) it promotes foreign DNA integration, (5) it enhances TopoIIα function, and (6) it promotes replication fork restart after stalling. It is important to develop small molecule inhibitors of Metnase because it promotes resistance to common cancer chemotherapeutics and promotes HIV integration. It represents a domesticated transposase: transposase biochemical activities that cause genome rearrangements and might predispose to malignancy have been subverted by primates to enhance DNA repair and genome stability. It is ironic that Metnase, instead of mobilizing DNA segments and moving them to alternative locations, promotes repair of free DNA ends. There are certainly unrecognized activities of Metnase, especially in its interactions with heterochromatin, where it might regulate unknown replication, transcriptional, or DNA repair processes. Given the wide activities of Metnase, one of the major goals of future investigation is to define whether there is a common mechanism for these activities, or whether they are based on distinct interacting partners.
One of the most intriguing features of Metnase is its presence solely in primates. Genome stability is certainly important for all organisms throughout evolution, and Metnase may have arisen late in response to increasing threats posed by invading transposons. There are certainly other possible explanations for its late arrival. For example, there is a mental retardation syndrome with a deletion at 3p26 near Metnase, and it is intriguing to speculate that Metnase may play a role in neuronal stability (Higgins et al. 2004). Other interesting questions are how can a protein that arose so recently interact with proteins involved in critical aspects of DNA dynamics and regulate so many important cellular functions? One answer derives from the fact that the SET and transposase domains of Metnase were each present long before they fused in primates. Thus, the separate domains probably evolved interaction partners independently, long before the gene fusion event. In this view, the fusion brought together distinct biochemical activities and interaction partners, yielding novel beneficial biochemical properties. Primate cells may have become addicted to Metnase function, selecting for its presence in those mammals. However, some thought must also be given to what was lost by this fusion. Since the SET domain of Metnase was present long before its fusion with the transposase, it is possible that losing this distinct SET activity might beneficially alter transcriptional regulation of gene expression during primate evolution. In like manner, the development of Metnase clearly altered the classic biochemical function of its transposase domain, which may have altered genomic stability in primates. Thus, the loss of specific activities by this fusion may have also played an important yet undiscovered role in primate development. In summary, Metnase is a unique transpose-derived protein that regulates several key cellular functions, and will therefore likely to continue to be a fertile area of study.
References
Ashworth A (2005) Oh what a tangled web it weaves: BRCA1 and DNA decatenation. Cancer Cell 8:95–97
Beck BD, Park SJ, Lee YJ, Roman Y, Hromas RA, Lee SH (2008) Human PSO4 is a Metnase (SETMAR) binding partner that regulates Metnase function in DNA repair. J Biol Chem 283:9023–9030
Bell O, Wirbelauer C, Hild M, Scharf AN, Schwaiger M, MacAlpine DM, Zilbermann F, van Leeuwen F, Bell SP, Imhof A, Garza D, Peters AH, Schubeler D (2007) Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J 26:4974–4984
Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, Schreiber SL, Lander ES (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128:669–681
Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127:1361–1373
Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123:581–592
Cordaux R, Udit S, Batzer MA, Feschotte C (2006) Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc Natl Acad Sci USA 103:8101–8106
DeHaro L, Wray J, Williamson E, Durant S, Corwin L, Gentry A, Osheroff N, Lee S-H, Hromas R, Nickoloff J (2010) Metnase promotes restart of stalled and collapsed replication forks. Nucleic Acids Res 38:2250–3500
Dreyfus DH (2006) The DDE recombinase: diverse roles in acquired and innate immunity. Ann Allergy Asthma Immunol 97:567–576
Edmunds JW, Mahadevan LC, Clayton AL (2008) Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 27:406–420
Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP (2004) Candidate genes for recessive non-syndromic mental retardation on chromosome 3p (MRT2A). Clin Genet 65:496–500
Hromas R, Wray J, Lee SH, Martinez L, Farrington J, Corwin LK, Ramsey H, Nickoloff JA, Williamson EA (2008) The human set and transposase domain protein Metnase interacts with DNA Ligase IV and enhances the efficiency and accuracy of non-homologous end-joining. DNA Repair 7:1927–1937
Kouzarides T (2007) Chromatin modifications and their function. Cell 23:693–705
Kriventseva EV, Koch I, Apweiler R, Vingron M, Bork P, Gelfand MS, Sunyaev S (2003) Increase of functional diversity by alternative splicing. Trends Genet 19:124–128
Kuraoka I, Ito S, Wada T, Hayashida M, Lee L, Saijo M, Nakatsu Y, Matsumoto M, Matsunaga T, Handa H, Qin J, Nakatani Y, Tanaka K (2008) Isolation of XAB2 complex involved in pre-mRNA splicing, transcription, and transcription-coupled repair. J Biol Chem 283:940–950
Lander ES et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Lee SH, Oshige M, Durant ST, Rasila KK, Williamson EA, Ramsey H, Kwan L, Nickoloff JA, Hromas R (2005) The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc Natl Acad Sci USA 102:18075–18080
Li J, Moazed D, Gygi SP (2002) Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem 277:49383–49388
Liu D, Bischerour J, Siddique A, Buisine N, Bigot Y, Chalmers R (2007) The human SETMAR protein preserves most of the activities of the ancestral Hsmar1 transposase. Mol Cell Biol 27:1125–1132
Mahajan KN, Mitchell BS (2003) Role of human Pso4 in mammalian DNA repair and association with terminal deoxynucleotidyl transferase. Proc Natl Acad Sci USA 100:10746–10751
McClendon AK, Rodriguez AC, Osheroff N (2005) Human topoisomerase II alpha rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J Biol Chem 280:39337–39345
Miskey C, Izsvak Z, Kawakami K, Ivics Z (2005) DNA transposons in vertebrate functional genomics. Cell Mol Life Sci 62:629–641
Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z (2007) The ancient Mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol 27:4589–4600
Plasterk RH, Izvsvak Z, Ivics Z (1999) The Tc1/Mariner superfamily of transposable elements. Trends Genet 15:326–332
Robertson HM, Zumpano KL (1997) Molecular evolution of an ancient mariner tansposon, Hsmar1, in the human genome. Gene 205:2003–2207
Roca J (2009) Topoisomerase II: a fitted mechanism for the chromatin landscape. Nucleic Acids Res 37:721–730
Roman Y, Oshige M, Lee Y-J, Goodwin K, Georgiadis MM, Hromas RA, Lee S-H (2007) Biochemical characterization of a SET and transposase fusion protein, Metnase (SETMAR) for its DNA binding and DNA cleavage activity. Biochemistry 46:11369–11376
Williamson EA, Farrington J, Martinez L, Ness S, O’Rourke J, Lee SH, Nickoloff J, Hromas R (2008a) Expression levels of the human DNA repair protein Metnase influence lentiviral genomic integration. Biochimie 90:1422–1426
Williamson EA, Rasila KK, Corwin LK, Wray J, Beck BD, Severns V, Mobarak C, Lee SH, Nickoloff JA, Hromas R (2008b) The SET and transposase domain protein Metnase enhances chromosome decatenation: regulation by automethylation. Nucleic Acids Res 36:5822–5831
Wray J, Williamson EA, Fnu S, Lee S-H, Libby E, Willman CL, Nickoloff JA, Hromas R (2009a) Metnase mediates chromosome decatenation in acute leukemia cells. Blood 114:1752–1758
Wray J, Williamson EA, Royce M, Shaheen M, Beck BD, Lee SH, Nickoloff JA, Hromas R (2009b) Metnase mediates resistance to topoisomerase II inhibitors in breast cancer cells. PLoS ONE 4:e5323
Zhang N, Kaur R, Lu X, Shen X, Li L, Legerski RJ (2005) The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J Biol Chem 280:40559–40567
Zhang J, Yu C, Pulletikurti V, Lamb J, Danilova T, Weber DF, Birchler J, Peterson T (2009) Alternative Ac/Ds transposition induces major chromosomal rearrangements in maize. Genes Dev 23:755–765
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shaheen, M., Williamson, E., Nickoloff, J. et al. Metnase/SETMAR: a domesticated primate transposase that enhances DNA repair, replication, and decatenation. Genetica 138, 559–566 (2010). https://doi.org/10.1007/s10709-010-9452-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9452-1