
Abstract
One hundred ninety-four germplasm accessions of fig representing the four fig types, Common, Smyrna, San Pedro, and Caprifig were analyzed for genetic diversity, structure, and differentiation using genetic polymorphism at 15 microsatellite loci. The collection showed considerable polymorphism with observed number of alleles per locus ranging from four for five different loci, MFC4, LMFC14, LMFC22, LMFC31 and LMFC35 to nine for LMFC30 with an average of 4.9 alleles per locus. Seven of the 15 loci included in the genetic structure analyses exhibited significant deviation from panmixia, of which two showed excess and five showed deficiency of heterozygote. The cluster analysis (CA) revealed ten groups with 32 instances of synonymy among cultivars and groups differed significantly for frequency and composition of alleles for different loci. The principal components analysis (PCA) confirmed the results of CA with some groups more differentiated than the others. Further, the model based Bayesian approach clustering suggested a subtle population structure with mixed ancestry for most figs. The gene diversity analysis indicated that much of the total variation is found within groups (H G /H T = 0.853; 85.3%) and the among groups within total component (G GT = 0.147) accounted for the remaining 14.7%, of which ~64% accounted for among groups within clusters (G GC = 0.094) and ~36% among clusters (G CT = 0.053). The analysis of molecular variance (AMOVA) showed approximately similar results with nearly 87% of variation within groups and ~10% among groups within clusters, and ~3% among clusters. Overall, the gene pool of cultivated fig analyzed possesses substantial genetic polymorphism but exhibits narrow differentiation. It is evident that fig accessions from Turkmenistan are somewhat genetically different from the rest of the Mediterranean and the Caucasus figs. The long history of domestication and cultivation with widespread dispersal of cultivars with many synonyms has resulted in a great deal of confusion in the identification and classification of cultivars in fig.
Similar content being viewed by others


Introduction
The fig, Ficus carica L., (Moraceae) is a classical fruit tree of antiquity associated with the beginning of horticulture in the Mediterranean basin (Zohary and Spiegel-Roy 1975). It is known to have been domesticated from a group of diverse spontaneous figs occurring in the south and east of the Mediterranean region sometime in the Early Neolithic period (Zohary and Hopf 1993). However, large fruited fig trees found in the deciduous forests of the Colchic district of northern Turkey and the Hyrcanic district of Iran and adjacent areas, which often intergrades into the Mediterranean figs, are considered by some botanists as a distinct ecotype of F. carica, and as a separate species, F. colchica Grossh. and F. hyrcanica Grossh., by others (Zhukovsky 1962). According to Vavilov (1951), Transcaucasia is considered as one of the centers of origin and diversity as one could see all phases of the domestication of fig in the southern Caucasus, where wild, transition, and modern fruit growing still exists.
The cultivated fig is gynodioecious, but is functionally dioecious, with pollination facilitated by the mutualistic interaction of pollinator wasps (Blastophaga psenes L.) between the two different fig types, Caprifig and edible fig (Kjellberg et al. 1987). The fig is an enclosed inflorescence that transforms into a hollow succulent receptacle called syconium. The syconium of female fig contains only long-styled pistillate flowers whereas that of the male (Caprifig) bears spongy, non-palatable syconia containing both staminate flowers and short-styled female flowers. Caprifig usually bears three crops: over wintering “mamme”, numerous “profichi” during spring, and “mammoni” during autumn. Figs are generally classified into Common, Smyrna, San Pedro, and Caprifig types mainly based on the floral biology and pollination behavior. Of the four types, Caprifig, although hermaphroditic, is functionally a male fig and is regarded as primitive while the Common-type, with only pistillate flowers developing into parthenocarpic fruits, is considered advanced and includes most commercial cultivars (Condit 1947). Smyrna and San Pedro types represent intermediate forms requiring pollination for normal fruit development with an exception of San Pedro type, which produces an early parthenocarpic crop mainly on older branches (Breba crop).
Domestication history and early migration along ancient trade routes have strong bearing on the modern distribution, genetic diversity and structure of fig. The discovery of carbonized figs in an early Neolithic site in the Jordan Valley, dating back 11,400–11,200 years ago, suggests that figs were first domesticated during the early Neolithic Revolution preceding cereal domestication (Kislev et al. 2006). As fig cultivation spreads to southern Arabia and subsequently into neighboring western Asia including Mesopotamia, Anatolia, Tanscaucasia, Persia, and other Middle-Eastern regions, introgression with local wild figs and landraces, and human selection, especially in Transcaucasia, resulted in recognition of numerous varieties and forms. Further westward migration of fig into Greece, Italy, Spain, Portugal and southward into Egypt added more cultivars. Spanish missionaries introduced fig into the New World in the mid-sixteenth century and North America soon thereafter. Franciscan missionaries were the first to plant figs in California sometime during the mid-ninteenth century and named the cultivar “Mission” (Condit 1955; Storey 1975). Further introductions of Smyrna and Caprifigs from France and Asia Minor occurred in the latter part of the century and the US Department of Agriculture (USDA) introduced the pollinator Blastophaga in 1890 (Condit 1947) to facilitate Smyrna fig production.
The long domestication history with numerous cultivars and further exchange and spread into other growing regions of the world has resulted in ambiguity in the description and nomenclature of fig cultivars. Condit (1955) listed more than 700 fig cultivars along with their synonyms and classified them into the four types, but a great deal of confusion still exists in the cultivar identification and their relationships. The lax use of cultivar names by growers and commercial nurseries, poor documentation of passport data during germplasm collection, substituting local and regional names for the same clonal cultivars, and existence of variants within cultivars are hindering proper identification and description of fig cultivars. It is critical for a germplasm repository to provide reliable information on the germplasm it holds and distributes through on-site evaluation. Characterization based on morphological criteria is often variable across years and locations due to the plasticity and susceptibility of these traits to genotype-environmental interactions. Molecular markers offer a stable and reliable alternative for genetic identification and characterization of germplasm collections. Recently, microsatellite, also known as Simple Sequence Repeats (SSR), randomly amplified polymorphic DNA (RAPD), inter-simple sequence repeat (ISSR), restriction length polymorphism (RFLP), and mitochondrial DNA RFLP markers have been used in fingerprinting, and assessing genetic diversity, structure and differentiation in fig collections (Khadari et al. 2001; Papdopoulou et al. 2002; Salhi-Hannachi et al. 2004; Khadari et al. 2005).
As part of an ongoing germplasm characterization effort at the USDA germplasm repository, at Davis, California, we analyzed a subset of the fig collection using microsatellite markers and the preliminary results are reported here. The study attempts to assess the genetic diversity and differentiation within the collection and elucidate the genetic relationships within and between different cultivar groups.
Materials and methods
Plant material, DNA extraction, and microsatellite analysis
About 194 fig accessions representing the four cultivar types and one accession each of F. palmata syn. pseudocarica and F. pumila were sampled from the germplasm collection maintained at the USDA National Clonal Germplasm Repository, Davis, California (Table 1). Total DNA was isolated using the CTAB method (Doyle and Doyle 1987) and further extracted with phenol–chloroform and treated with RNase to remove protein and RNA contaminants, respectively.
Sixteen microsatellite markers: MFC1 through MFC5 and MFC8 (Khadari et al. 2001); LMFC12, LMFC14, LMFC22, LMFC24, LMFC26, LMFC30, LMFC31, LMFC35, LMFC36, and LMFC37 (Giraldo et al. 2005) were PCR amplified separately in a 10-μl reaction mixture containing 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 2 mM MgCl2 (all included in 10 μl of 10× PCR buffer), 10 pmol of each primer, 200 μM of each dNTP, 2 U of Taq polymerase (Perkin Elmer Biosystems, California, USA), and 50 ng of template DNA. The PCR conditions were as follows: one cycle of 5 min at 94°C, 30 cycles of 94°C for 30 s, 55°C for 45 s, and 72°C for 1 min followed by one cycle of 7 min at 72°C. Amplified products were resolved using capillary electrophoresis on an ABI Prism 3100 Genetic Analyzer with the Data Collection software, version 1.2 (PE/Applied Biosystems). The data was further analyzed using Genescan, Version 3.1 and Genotyper, Version 2.5 and assembled as microsatellite genotypes as well as in binary format.
Data analysis
Population structure within fig germplasm collection
The marker, MFC5, which amplified two loci, was excluded from the computation of genetic diversity and differentiation parameters due to discrepancy in the assignment of alleles between two loci. However, the binary data from all 17 loci were used to compute the Nei and Li distance (Nei and Li 1979) based on the proportion of alleles shared between two accessions for all possible pair-wise combinations. The resultant matrix was subjected to a cluster analysis (CA) following the neighbor-joining (NJ; Saitou and Nei 1987) method to produce a phenogram. Bootstrap interior branch test (Dopazo 1994) was used to test the reliability of each interior branch on the tree. The multilocus SSR genotype data were pooled into groups based on the results of NJ cluster analysis and analyzed for various within-group genetic variability measures, such as mean number of alleles per locus and observed and expected levels of heterozygosities. Genetic divergence among groups was expedited using the distance Wagner procedure (Farris 1972 modified by Swofford 1981) based on a matrix of pair-wise distances between groups using Prevosti distance (Wright 1978).
Multivariate relationships among 142 fig accessions possessing unique multilocus genotypes were examined with principal components analysis (PCA) using the software NTSYSpc (Exeter Software). Accessions were projected along the first three principal axes to visualize genetic affinities.
Population sub-structuring within the fig germplasm collection (132 genotypes) was examined following a Bayesian clustering approach described by Pritchard et al. (2000), as implemented in the software package STRUCTURE 2.1 with different values for the number of clusters (K) ranging from 1 to 10. The burn-in length and number of MCMC reps after burn-in were set to 105 for each run with 10 replicates for each K to estimate the number of subpopulations. We choose the admixture model and the option of correlated allele frequencies between populations as suggested by Falush et al. (2003) in cases of subtle population structure. Two sets of runs were performed, one each with and without prior classification information from the CA. We let the degree of admixture alpha be inferred from the data. The parameter lambda, for allele frequency distribution, was set to unity, as suggested in the manual. STRUCTURE would attribute a probability Pr(X|K) given the data (X), and the log Pr (X|K) is used to determine the likely number of clusters (Pritchard et al. 2000). The K value that provides the maximum likelihood, called Ln P(D) in STRUCTURE, over the runs is generally considered as the most probable number of subdivisions. However, the interpretation of K should be treated with care as it merely provides an ad hoc approximation (Pritchard et al. 2000) and genuine and subtle population structure is likely to be missed by STRUCTURE. To find optimal alignments of independent runs, the computer program CLUMPP version 1.1 (Jakobsson and Rosenberg 2007) was used and the output obtained was used directly as input by the cluster visualization program DISTRUCT version 1.1 (Rosenberg 2004). A second order rate change with respect to K (ΔK) as defined in Evanno et al. 2005 was calculated to choose the optimum number of clusters K.
Genetic diversity within and among groups
Accessions with unique multilocus fingerprints were grouped according to the results of the CA and the genotypic data matrix was subjected to various within groups genetic diversity measures, such as mean number of alleles/locus, polymorphic index, and observed and expected levels of heterozygosity. The fixation index (F; Wright 1965), which is equal to (H exp − H obs)/H exp, where H exp and H obs refer to expected and observed heterozygosity, respectively, was computed for all loci and the significance of which was tested using Fisher’s Exact Test as described by Guo and Thompson (1992) and implemented in the software Arlequin (Excoffier et al. 2005).
A hierarchical gene diversity analysis following the method described by Nei (1973, 1978) was performed considering relevant levels of population subdivision: groups and clusters. The total gene diversity (H T) is partitioned into its components so that
HT = HG + DGC + DCT, where HG is average gene diversity within groups, DGC is gene diversity between groups within clusters and DCT corresponds to diversity between clusters. The relative importance of the three components are expressed in terms of coefficient of gene differentiation (G) values (G G , GGC, GCT) that are obtained from the ratios of each component to HT. All computations were performed on an Excel spreadsheet.
Results
Allelic variation and genetic relationship among fig cultivars
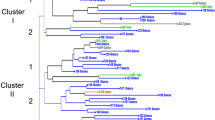
The fig collection examined showed considerable polymorphism with observed number of alleles per locus ranging from four for MFC4, LMFC14, LMFC22, LMFC31 and LMFC35 to nine for LMFC30 with an average of 4.9 alleles per locus (Table 2). The observed and expected levels of heterozygosity for different loci indicated that the gene and genotype frequencies in fig significantly deviated from the Hardy–Weinberg expectations for seven of fifteen loci included in the analysis. Of the seven loci exhibiting deviation, two showed excess and five showed deficiency of heterozygotes. However, the mean heterozygosity across loci conformed to panmixia. Comparison of multilocus genotypes revealed many instances of apparent synonymy or misidentification of cultivar names. The CA using the neighbor-joining method revealed ten groups (Fig. 1) and subgroups were evident within each of them. Interestingly, the groups contained an assortment of Smyrna, Common, and San Pedro type figs interspersed with occasional Caprifigs, suggesting that the sex expression in fig is simply inherited and the different fig types share a common gene pool. The CA unraveled 32 instances of synonymy at least involving two cultivars. In most cases the cultivars within the synonymous groups showed a high degree of morphological similarity with respect to tree architecture, vegetative and pomological characteristics.

Neighbor-joining tree showing the genetic relationships among the fig genotypes included in the study (support for branches is based on bootstrap interior-branch test; Dopazo 1994)
Groups 1 and 2 mostly consisted of Common fig, except for a few San Pedro and a single Caprifig, with nine instances of synonymy. For example, figs with different names but identical multilocus fingerprint included: (1) six greenish-yellow figs with strawberry-yellow flesh inlcuded ‘Lemon’, ‘Dokkar’, ‘L.S.U. Everbearing’, ‘White Texas Everbearing’, and ‘Trojano’ characterized by a widely grown cultivar ‘Kadota’; (2) five greenish-yellow to light brown skinned figs with light amber to yellow colored pulp included ‘Archipel’, ‘Malcolm’s Super Giant’, ‘Drap D’or’, and ‘Encanto Brown Turkey’ with two other closely related cultivars, ‘Alma’ and ‘Golden Celeste’; (3) three medium sized purple/black figs ‘Vista’, ‘Violette de Bordeaux’, and ‘Beers Black’; (4) two green figs with amber flesh ‘Verte’ and ‘Calverte’; and (5) three green figs with thick rind with light strawberry flesh ‘Paradiso’, ‘Monstrueuse’, and ‘Ischia Green.’
Group 3 again predominantly contained Common figs and showed three instances of synonymy. A purple/black fig assemblage consisted of ‘Sal’s fig’, ‘Dark Portuguese’, and ‘Abruzzi’ with dark strawberry flesh. A second yellow/green fig assemblage contained cultivars with amber flesh, ‘Genoa White’, ‘Genoa’, and ‘Harvey Adriatic.’ A third group with yellow/green skin and amber to light strawberry red colored flesh included ‘Brunswick’, ‘Rattlesnake Island’, ‘Capitola Long’, ‘Doree’ and an amber to light purple skinned fig, ‘Red Italian’ and, according to Condit, these cultivars are similar to each other (Condit 1955).
Group 4 is exclusively made up of Common figs and contained two instances of synonymy. One consisted of two light to medium purple figs with amber flesh ‘Santa Cruz Dark’ and ‘Giant Amber’, and a second group of three elongated, pear-shaped, dark purple/black figs, ‘Noire de Caromb’, ‘Cuello Dama Negro’, and ‘Charles Allen’, appeared to be closely genetically related to the popular cultivar, ‘Mission.’
Group 5 is made up of Common figs with two occurrences of synonymy, one of which contained six genotypes and is characterized by a widely grown purple fig with strawberry flesh, ‘Brown Turkey’, and its well known synonyms, ‘California Brown Turkey’, ‘Braun Turkey’, ‘Black Jack’, and two other purple cultivars with strawberry flesh similar to ‘Brown Turkey’ matching closely in fruit traits to ‘Walker’ and ‘Blue Giant.’ A second one consisted of two green figs with amber flesh, ‘Catarulla’ and Green Italian.’
Groups 6, 7, 8, 9, and 10 showed several instances of synonymy involving two accessions in each of these occurrences, except for one in group 9 with four green figs with amber flesh, ‘Deanna’, ‘UCR278’, ‘Orphan’, and ‘Algerian Watts.’ Interestingly, a subgroup in group 8 contained several cultivars from Turkmenistan, ‘Shevlan 1’, ‘Shevlan 2’ and ‘Shevlan 3’, ‘Caucasus 3’, ‘Caucasus 6’, ‘Nuhurskii’, ‘Shih Berdy’, ‘Zheltyi from Seidoc’, ‘Inzhyr from Sopyev’, ‘Akinzhyr Kuruzhdeiskii’, ‘Kury Gol’, and ‘Kugitangskii Chernyil’. Group 9 is predominantly made up of several selections, such as ‘Tena’, ‘UCR 291’, ‘Conadria’, Gulbun Selection’, ‘Jurupa’, ‘Flanders’, ‘UCR 271’, ‘Deanna’, ‘UCR278’, ‘Orphan’ and a number of parental genotypes used in Condit’s breeding program at the University of California, Riverside.
The PCA confirmed the groups recognized in the CA. The first three principal axes accounted for only 16.4% of the total variation, indicating the multidimensional nature of the molecular variation in fig. All three principal axes were important to visualize the marginal differentiation among groups recognized in the CA (Fig. 2). Groups 8 and 10 were clearly resolved along the third axis while groups 1, 2, 3 and 4 are somewhat overlapping, but showed subtle differentiation in the 3D-space along the first three axes. Groups 5, 6, 7 and 9 were somewhat intermediate, found at the center connecting the rest of the groups.

Three dimensional projection of fig accessions along the first three principal axes accounting for a total of 16.4% of the total molecular variation. Groups are based on the cluster analysis using neighbor-joining method (see Fig. 1)
Genetic diversity and population structure in fig
Measures of within-group genetic diversity are summarized in Table 3. The groups differed significantly with respect to frequency and composition of alleles for different loci. While most clusters possessed two or more moderate to high frequency alleles, there are many low frequency, group specific alleles. The mean number of alleles per locus ranged from 2.5 for group 6 to 4 for group 8 with an average of 3 alleles per locus. Percentage of polymorphic loci ranged from 86.7% for groups 6, 7 and 9 to 93.3% for the remaining groups 1–5, 8, and 10 with an average 91.3%. The mean observed heterozygosity levels were consistently higher than the expected panmictic proportions, except for groups 3 and 8, which showed marginally lower levels. The mean observed heterozygosity ranged from 0.450 for groups 8 to 0.665 for group 1 with an average of 0.537. The fixation index indicated deficiency of heterozygotes within each of the groups as compared to Hardy–Weinberg expectations except for groups 3 and 8. Fisher’s Exact Test suggested that the groups differentiated significantly with respect to frequency and pattern of distribution of alleles at different loci assayed.
Further analysis of genetic divergence among the ten groups identified by the CA using the distance Wagner procedure with midpoint rooting option produced three distinct clusters (Fig. 3). Cluster 1 contained groups 1, 2, 3, 4 and 6, cluster 2 had groups 5, 9 and 10, and cluster 3 included groups 7 and 8.

Distance Wagner tree showing the genetic relationships among groups identified in the cluster analysis. Groups are based on the cluster analysis using neighbor-joining method (see Fig. 1)
The gene diversity analysis based on allele frequencies for the 10 groups obtained by the NJ cluster analysis (Table 4) excluding the duplicate accessions indicated that the total gene diversity (H T), a measure of mean heterozygosity in the total collection, is reasonably high across loci, ranging from 0.269 for LMFC24 to 0.735 for MFC1 with an average of 0.514. The intragroup gene diversity summed over all loci accounted for a significant portion of the total diversity (H G/H T = 0.853; 85.3%) and the remaining 14.7% of the total diversity averaged across loci is due to differentiation among groups (G GT = 0.147). The coefficient of gene differentiation (G GT) varied across loci as it is highly dependent on the total genetic diversity (H T) and may produce larger values although gene differentiation is small. Further hierarchical partitioning of the G GT component into groups within and among clusters revealed by the distance Wagner tree indicated that a greater proportion of gene diversity among groups resides in the among groups within clusters component (G GC = 0.094; 9.4%) compared to diversity due to differentiation among clusters (G CT = 0.053; 5.3%). The hierarchical partitioning of allele frequency variation in the total population across loci into components due to differentiation within and among groups and within and among clusters using the nested AMOVA procedure (Table 5) produced results similar to gene diversity analysis. The pattern of distribution of diversity indicated that ~87% of total variation was accounted for within-group, ~10% for genetic differentiation among groups within cluster, and 2.9% for differentiation among clusters.
Bayesian analysis of population structure with and without incorporating prior population information based on the CA revealed different clustering results as compared to distance based analysis. The estimated likelihood value (Ln Pr X|K) attained a maximum value around K = 4 and K = 5 (Fig. 4b), beyond which the likelihood values declined and the standard deviation associated with the estimates has increased (Fig. 4). However, the same analysis, when prior population information based on the CA was incorporated to facilitate estimation of K, the Ln Pr(X|K) estimate reached the highest value around K = 7 and K = 8 (Fig. 4a), beyond which the estimates varied considerably among the replicate runs resulting in significant standard deviation. In general the Bayesian estimates of K tend to be somewhat lower than the number of subdivisions observed in a distance based CA using the neighbor-joining algorithm. The ad hoc quantity based on the second order rate of change of the likelihood function (∆K) did show two equivalent peaks at K = 2 and K = 5, and a lesser one at K = 7 for the runs with prior population information (Fig. 4c), while there was one clear peak at K = 2 and a lesser peak around K = 4 and K = 5 for the runs without prior information (Fig. 4c). Cluster visualization for the permuted average Q-matrix generated by CLUMPP for the runs of STRUCTURE associated with the average maximum likelihood values are presented in Fig. 5. The runs with K = 5 appears to be the best to model the fig variability for both with or without prior population information. Generally, taking into account the population information generated by the CA did not help in resolving the genetic structure using the Bayesian approach. In other words, there is some level of incongruence in the assessment of genetic structure between the distance and model based approaches. At K = 5 without prior population information, the STRUCTURE analysis produced results somewhat matching the multivariate relationships revealed by the CA and the PCA. Overall, the distance based CA, the PCA and the model based CA examined in this study suggest mild genetic structure and most individuals have mixed ancestry.

Graphs a and b: Mean posterior probabilities, Ln P(D), averaged across 10 runs, for the data set comprising 132 fig genotypes, for different K ranging from 1 to 10, considered for the estimation of the number of subpopulations, with and without consideration to the number of groups identified in the distance based cluster analysis, respectively. Graphs c and d: Second order rate change with respect to K (∆K) as a function of K computed based on the posterior probabilities averaged across 10 replicates with and without considerations to the number of groups identified in the distance based cluster analysis, respectively

Inferred population structure for K = 2, K = 5, and K = 7 as the probable numbers of subpopulation with the fig germpasm collection consisting of 132 fig genotypes. Each individual is represented by a thin tine partitioning into K colored segments representing the membership fraction in K clusters. Plots generated with DISTRUCT (Rosenberg 2004), is based on the Q-matrix consensus permuted across 10 replications for each K using CLUMPP (Jakobsson and Rosenberg 2007)
Discussion
Molecular characterization and genetic identification of cultivars
Knowledge of genetic diversity, population structure and differentiation significantly contributes to effective conservation, management and utilization of germplasm collections. Genetic characterization of ex situ collections offers insight into the amount and patterns of distribution of genetic diversity and permits classification of germplasm based on genetic similarities and differences. Nowadays, gene banks around the world are focusing on genetic and phenotypic characterization of germplasm collections in order to promote efficient utilization of germplasm in breeding and development of crops. Characterization permits identification of deficiencies in collections and planning for future collection efforts to strategically enrich existing collections. In clonally propagated, perennial species such as fig, germplasm accessions are preserved as unique genotypes, the genetic and phenotypic integrity of which is important for breeders and researchers who look for particular combinations of traits or genes in an accession. Phenotypic analysis of variation in clonally maintained, perennial crop collections are age and management-dependent and subject to genotype-environmental interactions and consequently not comparable across environments, but biomolecular evaluations offer a comparable measure of genetic diversity and establish the identity for individual accessions.
Germplasm collections of most clonally propagated species often contain morphologically similar accessions having different genetic and geographic origins. Further, genetically identical cultivars may have different names in different collections and countries, probably due to lax use of names by growers, nurserymen, and traders, corruption in English transliteration of original names, the presence of variants within cultivars, and lack or poor documentation of passport data. Fig is adapted to a wide range of climates from tropical, subtropical, Mediterranean, and even to temperate conditions, and has a long domestication and cultivation history, which has led to recognition of numerous ecotypes and landraces selected and maintained by indigenous people for their adaptation to local environments and farming systems, and subtle fruit qualities, which generally possess local synonymous names, a problem that plagues germplasm collections of clonal crops (Galet 1990; Lebot and Aradhya 1991; Aradhya et al. 1995). Condit (1955), in his monograph on fig varieties, lists more than 700 cultivars and the majority have large numbers of synonyms. Many of the old and popular cultivars such as ‘Kadota’, ‘Brown Turkey’, ‘Ischia Green’ and ‘Brunswick’ often possess several synonyms and they generally possessed similar tree structure, morphology and fruit characteristics. Deciphering genetic identity and relationships among these cultivars is complicated due to occurrence of extensive synonymy and non availability of authentic source cultivars for comparison. The genetic and geographic origin of most of these cultivars is unknown and associated passport data are incomplete, inaccurate, or missing in most germplasm collections.
The figs from Turkmenistan have allied tightly in group 8 with good bootstrap support, indicating some level of differentiation from the rest of the figs. However, group 8 also contained ‘Zidi’, a dark purple Smyrna fig from Morocco, ‘Calimyrna’, a yellow Smyrna fig commercially grown in California, and two other U.S. cultivars, ‘Snowden’ and ‘Osborne Prolific.’ This group may represent non-Mediterranean type wild figs found in the Hyrcanic regions of the south Caspian Sea, which some botanist treat as a separate species, F. hyrcana (Zhukovsky 1962). Both Mediterranean and non-Mediterranean wild figs are fully interfertile and produce hybrids that are adapted to a wide range of ecological conditions (Storey and Condit 1969). Group 9 features cultivars developed in the early California breeding program (Condit 1947; Storey 1975) such as ‘Conadria’, ‘Deanna’, ‘Tena’, ‘Jurupa’, ‘Gulbun’ and ‘Flanders’ and some of the cultivars used in the hybridization program. The cultivar ‘Adriatic’, which has been extensively used in the California fig breeding program, also clusters within this group. The cultivars ‘Brunswick’, ‘Rattlesnake’, and ‘Capitola Long’ showed identical multilocus genotypes, and were shown to be identical in an earlier study based on sequence-related amplified polymorphisms (Gao and Quiros, unpublished). Overall, the classification of fig cultivars is largely based on skin and pulp color, floral biology, pollination behavior and parthenocarpy, which are probably governed by simple Mendelian genes and may be unrelated to molecular markers.
Genetic diversity and population structure
The fig germplasm collection harbors moderate to high levels of genetic polymorphisms across the microsatellite loci assayed with 140 unique multilocus genotypes out of a total of 194 accessions included in the study. The heterogeneity among loci for levels of heterozygosity and fixation index reflects a complex selection history and genetic structure of populations from which the fig cultivars were originally selected. Averaging over loci, the fig collection approaches panmixia, although some loci deviated significantly, indicating differential selection among loci. The mild genetic structure within the fig collection with deeply dissected branches on the phenetic tree suggests that most variation is locked up at the level of individuals as polymorphic, multilocus heterozygotes. The ten groups identified based on the cluster analysis contained an assortment of fig types, Smyrna, Caprifig, San Pedro, and Common, indicating shared ancestry or evolutionary background and are connected through gene flow via Caprifig, the main pollen source (Condit 1947). However, support for some groups was marginal, indicating the complex multidimensional nature of molecular variation.
The weak genetic structure observed in the present study is probably due to the fact that fig circulates genetic variability across different fig types through a dynamic mutation-recombination process facilitated by a complex pollination mechanism involving the symbiotic relationship between the fig and its pollinator. Further, the genetic relationships within and among fig groups observed in the CA should reflect a complex combination of natural evolution, genetic drifts and founder events during domestication, historical migration of cultivars along human migrations from the center of origin and diversity to secondary centers and regions of commercial production, and genetic modifications through modern plant breeding. Although there was marginal evidence for differentiation, there was marked differences among genetic groups with respect to composition and frequency of alleles for different loci as indicated by the Fisher’s Exact Test. Clonally propagated perennial species such as fig are known to carry relatively high genetic load and tend to exhibit an excess of heterozygotes as a mechanism to overcome the deleterious effects of recessive mutations (Klekowski 1988).
Prevalence of Common fig in cultivation around the world probably indicates that human selection has historically favored parthenocarpic fig over pollination dependent Smyrna and San Pedro types, especially in regions lacking the pollinator wasp. Parthenocarpy in Common fig was probably selected early in the domestication history dating back to the early Neolithic period (Kislev et al. 2006) and possibly derived as a point mutation favored by humans. On the contrary, Lev-Yadun et al. (2006) points out that Common figs are dioecious, with male trees producing inedible seedless figs that maintain the pollinating wasps. It is known that the parthenocarpic Common fig, if pollinated, does produce better quality figs than parthenocarpically developed figs, and contain viable seeds, the progeny of which segregates into male and female figs.
Both Bayesian and distance based approaches used to examine the genetic structure and differentiation revealed weak genetic structure, probably due to inherently narrow genetic base from which the fig was domesticated, combined with historical migration of germplasm and the outcrossing mode of pollination, which have countered human selection in different fig growing regions of the world. Nevertheless, the CA using the neighbor-joining method identified ten, somewhat narrowly differentiated groups, some of which were further confirmed in the PCA analysis. The Bayesian analysis indicated that most fig genotypes have mixed ancestry, which becomes clear as the K value incraeses in the analysis. At K = 5 without prior population information, the simulation attained the highest likelihood value and had the higher clusteredness, while the cluster composition and membership coefficients somewhat reflected within and among group relationships in the CA and PCA. Although members from different clusters revealed by the CA moved around a bit among different Bayesian clusters at K = 5, most members from groups 1, 2 and 3 formed a cluster, members predominantly from groups 4, 5, 6 and 7 formed a second cluster, and most members from groups 9 and 8 came together in a cluster. However, some members from group 9 were found scattered among three different clusters and similarly some of 6 and 7. Most of the members of group 10 formed a cluster, some showing affinity with members of group 7.
Overall, it is challenging to infer the genetic structure and differentiation of outcrossing plant species such as fig with long history of domestication, extensive dispersal, and wide range of adaptation. Most of the alleles are widespread and formed gene frequency clines within and among groups and clusters reflecting the biogeographic history of fig. Further, as warned by the authors of STRUCTURE, the method of inferring K is an ad hoc procedure based on a set of uncertain assumptions and that the inferred K may not always have a clear biological interpretation (Pritchard et al. 2000). Even the ∆K approach based on the rate of change in the log probability of data between successive K values (Evanno et al. 2005) could not accurately predict the K in fig.
Genetic diversity within and among groups
Organization of genetic diversity in clonally propagated species germplasm collections is reminiscent of historical genetic structure originating from the complex interaction of evolutionary forces and domestication history of the species. The gene pool of fig examined possesses significant genetic variability and exhibits narrow differentiation among the ten genetic groups identified by the CA and PCA. However, the model based cluster analysis indicated that most fig genotypes had mixed ancestry and molecular variation is clinal without clear differentiation. The geographic or genetic basis for relationships among cultivars within and among groups and clusters is difficult to decipher due to lack or incomplete passport data. However, groups differed for the composition and frequency of alleles for different loci indicating some sort of mild sub-structuring within the collection. Earlier studies have demonstrated clustering of fig genotypes on a geographic basis (Papdopoulou et al. 2002; Salhi-Hannachi et al. 2006), but on limited sampling basis. Fig being a functionally dioecious, there is extensive species-wide gene flow within and among different groups and fig types, and the subtle substructure noticed in this study probably reflects a complex combination of effects of historical dispersal of cultivars and human selection. Further, the weak genetic structure of fig is probably suggestive of a single, complex gene pool featuring extensive dispersal of cultivars homogenizing the local populations. Earlier studies in fig generally reported increased effective population size reducing population subdivision with most variation tending to be within populations (Khadari et al. 1995; Papdopoulou et al. 2002; Giraldo et al. 2005). However, a study based on mtDNA restriction fragment length variation demonstrated slightly higher levels of differentiation among natural population (G ST = 0.323) and among groups of populations (G ST = 0.284) of fig from the Mediterranean region (Khadari et al. 2005).
Overall, the gene pool of cultivated fig analyzed possesses substantial genetic polymorphism and exhibits narrow differentiation. It is evident that fig accessions from Turkmenistan are somewhat genetically different from the rest of the Mediterranean and the Caucasus figs. A long history of domestication and cultivation with extensive dispersal of cultivars has often resulted in a great deal of confusion in the identification and classification of cultivars.
References
Aradhya MK, Zee FT, Manshardt RM (1995) Isozyme variation in lychee (Litchi chinensis Sonn). Sci Hortic 63:21–35
Condit IJ (1947) The fig. Chronica Botanica Co., Waltham
Condit IJ (1955) Fig varieties: a monograph. Hilgardia 23:323–538
Dopazo J (1994) Estimating errors and confidence intervals for branch lengths in phylogenetic trees by bootstrap approach. J Mol Evol 38:300–304
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Farris JS (1972) Estimating phylogenetic trees from distance matrices. Am Nat 106:645–668
Galet P (1990) Cépages et Vignobles de France Tome II L’Ampélographie FranHaise, 2nd edn. Imprimerie Charles Dehan Press, Montpellier
Giraldo E, Viruel MA, Lopez-Corrales M, Hormaza JI (2005) Characterization and cross-species transferability of microsatellites in the common fig (Ficus carica L.). J Hort Sci Biotechnol 80:217–224
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Khadari B, Lashermes P, Kjellberg F (1995) RAPD fingerprints for identification and genetic characterization of fig (Ficus carica L.) genotypes. J Genet Breed 49:77–86
Khadari B, Hochu I, Santoni S, Kjellberg F (2001) Identification and characterization of microsatellite loci in the common fig (Ficus carica L.) and representatives species of the genus Ficus. Mol Ecol Notes 1:191–193
Khadari B, Grout C, Santoni S, Kjellberg F (2005) Contrasted genetic diversity and differentiation among Mediterranean populations of Ficus carica L.: a study using mtDNA RFLP. Genet Resour Crop Evol 52:97–109
Kislev ME, Hartmann A, Bar-Yosef O (2006) Early domesticated fig in the Jordan Valley. Science 312:1372–1374
Kjellberg F, Gouyon PH, Ibrahim M, Raymond M, Valdeyron G (1987) The stability of the symbiosis between dioecious figs and their pollinators: a study of Ficus carica L. and Blastophaga psenes L. Evolution 41:693–704
Klekowski EJ (1988) Genetic load and its causes in long-lived plants. Trees, 195–203
Lebot V, Aradhya KM (1991) Isozyme variation in taro [Colocasia esculenta (L.) Schott] from Asia and the Pacific. Euphytica 56:55–66
Lev-Yadun S, Ne’eman G, Abbo S, Flaishman MA (2006) Comment on “Early domesticated fig in the Jordan Valley”. Science 314:1683
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M, Li W (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Papdopoulou K, Ehaliotis C, Tourna M, Kastanis P, Karydis I, Zervakis G (2002) Genetic relatedness among dioecious Ficus carica L. cultivars by randomly amplified polymorphic DNA analysis, and evaluation of agronomic and morphological characters. Genetica 114:183–194
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Salhi-Hannachi A, Mokhtar T, Salwa Z, Jihene H, Messaoud M, Abdelmajid R, Mohamed M (2004) Inter-simple sequence repeat fingerprints to assess genetic diversity in Tunisian fig (Ficus carica L.) germplasm. Genet Resour Crop Evol 51:269–275
Salhi-Hannachi A, Chatti K, Saddoud O, Mars M, Rhouma A, Marrakchi M, Trifi M (2006) Genetic diversity of different Tunisian fig (Ficus carica L.) collections revealed by RAPD fingerprints. Hereditas 143:15–22
Storey WB (1975) Figs. In: Janick J, Moore JN (eds) Advances in fruit breeding. Purdue University Press, Purdue, pp 568–589
Storey WB, Condit IJ (1969) Fig (Ficus carica L.). In: Ferwerda FP, Wit F (eds) Outlines of perennial crop breeding in the tropics, Misc paper 4. Landbouwhogeschool, pp 259–267
Swofford DL (1981) On the utility of the distance Wagner procedure. In: Funk VA, Brooks DR (eds) Advances in cladistics: proceedings of the first meeting of the Willie Hennig Society, New York Botanical Garden, New York, pp 25–43
Vavilov NI (1951) The origin, variation, immunity and breeding of cultivated plants. The Ronald Press Company, New York
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420
Wright S (1978) Evolution and the genetics of populations, vol. 4. Variability within and among populations. University of Chicago Press, Chicago
Zhukovsky PM (1962) Cultivated plants and their relatives (abridged translation by Hudson PS). Commonwealth Agricultural Bureaux, England
Zohary D, Hopf M (1993) Domestication of plants in the old world. Clarendon Press, Oxford
Zohary D, Spiegel-Roy P (1975) Beginnings of fruit growing in the old world. Science 187:319–327
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Aradhya, M.K., Stover, E., Velasco, D. et al. Genetic structure and differentiation in cultivated fig (Ficus carica L.). Genetica 138, 681–694 (2010). https://doi.org/10.1007/s10709-010-9442-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9442-3