
Abstract
Hyperphosphatemia has been associated with increased mortality in chronic kidney disease but the nature of such a relation in the general population is unclear. To investigate the association between phosphate (P) levels and all-cause and cause-specific mortality, we assessed two cohorts from the Rotterdam Study, with follow-up of 14.5 (RS-I) and 10.9 (RS-II) years until January 2012 with availability of fasting phosphate levels. Deaths were classified according to International Classification of Diseases into 7 groups: cardiovascular, cancer, infections, external, dementia, chronic lung diseases and other causes. Sex-stratified Weibull and competing-risks models were adjusted for age, BMI and smoking. Hazard ratios are expressed per 1 mg/dL increase in phosphate levels. The total number of participants included 3731 (RS-I, 2154 women) and 2494 (RS-II, 1361 women) subjects. The main outcome measures were all-cause and cause-specific mortality. A significant positive association was found between phosphate and all-cause mortality in men (pooled HR (95% CI): 1.46 (1.26–1.69)) but not in women (0.90 (0.77–1.05)). In men, higher phosphate increased the risk for cardiovascular mortality (1.66 (1.29–2.14)), other causes (1.67 (1.16–2.40)) and chronic lung disease mortality (1.94 (1.02–3.72)), the latter driven by mortality due to chronic obstructive pulmonary disease (COPD) (4.44 (2.08–9.49)). No relations were found for mortality due to infections, cancer, dementia or external causes. In conclusion, serum P is associated with increased all-cause, cardiovascular and COPD mortality in men but not women. The association with COPD mortality is novel and needs further research on underlying mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphorus is the sixth most common element in the human body and the second mineral in abundance [1]. It plays an important structural role in hard tissues, such as bone, and exerts critical regulatory roles in metabolic and signaling pathways [1].
The majority of phosphorus is stored in bone (85%) where it is complexed with calcium in the form of hydroxyapatite, whereas 15% of phosphorus is located in the intracellular compartment while less than 1% is present in extracellular fluids. In blood, phosphorus exists in two main forms: a) an organic form bound to proteins (70%), b) an ionized form (30%), known as inorganic phosphorus, or phosphate, that circulates freely [1].
Traditionally, phosphate homeostatic mechanisms have been ascribed to the actions of parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) [1, 2]. Recently, an equally important new axis of phosphate regulators was discovered [2, 3], composed of the so-called phosphatonins: fibroblast growth factor 23 (FGF23), synthesized mainly in osteocytes, and its co-receptor α-Klotho [3, 4]. The FGF23/α-Klotho axis increases Purinary excretion [5].
Monogenic disorders causing extreme phosphate concentrations are associated with rickets in severe hypophosphatemia and calcinosis in severe hyperphosphatemia [5]. Recently, milder hyperphosphatemia was shown to increase cardiovascular mortality in chronic kidney disease (CKD) [6]. Subsequently, this association was reported also in non-CKD population [7,8,9,10]. Interestingly, sex differences have been described with associations found in men but not women for all-cause mortality and subclinical atherosclerosis [9]; the underlying reasons are not understood. In addition to serum phosphate levels (P), high P intake has recently been found to increase mortality [11].
The objectives of this study were to assess the association of P with all-cause and, in detail, cause-specific mortality within two cohorts of the population-based Rotterdam Study, and to test for potential sex differences in these associations.
Materials and methods
Study population
The Rotterdam Study is a prospective study of men and women designed to investigate the incidence and determinants of chronic disabling diseases. Rationale and design has been described elsewhere [12]. This research was performed in two cohorts within the Rotterdam Study, the Rotterdam Study I cohort (RS-I), initiated in 1990 in 7983 subjects, and the Rotterdam Study II cohort (RS-II) initiated in 2000 in 3011 subjects. All participants were 55 years or more at recruitment and have been assessed at baseline and through several follow-up visits. P was measured in the non-fasting state at baseline visit of RS-I (referred to as RS-I-1) and in the fasting state at the second follow-up visit of RS-I (RS-I-3, referred to as RS-I) and the baseline visit of RS-II (Fig. 1). The fasting state may modify the association between P and mortality [10]. Therefore, our main analysis was based on data from RS-I-3 and RS-II because P was assessed in the fasting state; subsequently we checked if the observed results followed similar patterns in RS-I-1, where non-fasting samples are available. A total of 3731 participants from RS-I and 2494 from RS-II were included for these analyses, all of them with signed informed consent and available phosphate levels. The Rotterdam Study was approved by the institutional review board (Medical Ethics Committee) of the Erasmus Medical Center and by the review board of the Netherlands Ministry of Health, Welfare and Sports. The approval has been renewed every 5 years.

Flowchart for time line, design and sample size for the analyses
Laboratory measurements
The amount of phosphorus determined in blood corresponds to the inorganic fraction, or phosphate (mg/dL), assessed with a method based on the formation of ammonium phosphomolybdate [1].
Total calcium determination (mg/dL) was done through a colorimetric o-cresolphthalein complex one method (Merck Diagnostica, Amsterdam, The Netherlands, for RS-I-1; and Roche, Mannheim, Germany, for RS-I and RS-II). Levels of 25-hydroxyvitamin D (nmol/L) were determined through an electrochemiluminescence immunoassay. We applied cosinor regressions to adjust 25-hydroxyvitamin D for season and year. After testing for seasonality applying the dickey fuller test, we proceeded to perform a time transformation on sine and cosine terms (sin(2*pi*time/12)). Afterwards, we proceeded to regress the serum vitamin D levels on those terms to get the mesor, that is, the mean value of the cosinor regression. We then computed the difference between the mean of each season and the mesor, and adjusted every individual value accordingly [13, 14]. Levels of 1,25-dihydroxyvitamin D3 were assessed in a subset of participants from RS-I-1 through 125I-radioimmunoassay (IDS, Boldon, UK). Creatinine was determined through a sarcosine-based colorimetric assay and standardized against isotope dilution mass spectrometry (ID-MS). Cystatin C was assessed through particle enhanced immunoturbidimetric assay. C-reactive protein (CRP) levels were measured through an agglutination method with antibodies. Magnesium (Mg) levels were determined with a colorimetric method based on xylidyl blue. Glucose and cholesterol levels were determined by standard enzymatic methods [12].
Covariates
We assessed the distribution of potential confounders across P quintiles, such as age, body mass index (BMI), smoking status, calcium, 25-hydroxyvitamin D levels, creatinine, estimated glomerular filtration rate (eGFR), C-reactive protein (CRP), glucose, magnesium, total cholesterol to HDL cholesterol ratio and prevalent diabetes mellitus and cardiovascular disease (CVD). BMI, smoking status, prevalent diabetes mellitus and prevalent CVD were assessed as previously described [12]. The diagnoses of prevalent and incident chronic obstructive pulmonary disease (COPD) cases was based on an obstructive pre-bronchodilator spirometry (FEV1/FVC < 0.7), according to GOLD guidelines [15]. P intake at baseline visit (RS-I-1) was collected in a subsample of participants through a validated semiquantitative food frequency questionnaire. The Chronic Kidney Disease Epidemiology Collaboration equations based on creatinine [16, 17] were applied to estimate eGFR (mL/min). Additionally, cystatin C-based eGFR was estimated for subjects with creatinine-based eGFR less than 60 mL/min, as previously recommended [16].
Assessment of all-cause and cause-specific mortality
Information on vital status is obtained continuously from the municipal authorities in Rotterdam. The cohorts are monitored for mortality through computerized linkage of the study database to medical files of general practitioners. Two research physicians independently coded the mortality events according to ICD-10. Medical specialists in the respective field reviewed and confirmed the diagnosis. Information on cause-specific mortality was available until January, 2012.
Different causes of mortality were recorded according to ICD-10 codes and firstly grouped into cardiovascular diseases (CVD), cancer and other causes. To perform comprehensive analyses, the group of other causes was further categorized into external causes, dementia, infections, chronic lung disease and other causes in the strict sense, as previously described [18].
Statistical analysis
Subjects with fasting P measurements from RS-I and RS-II were analyzed separately and in a meta-analysis. Additionally, we analyzed subjects with non-fasting P from RS-I-1.
Due to sex differences in P [19] and in its association with health outcomes [9], we built sex-stratified analyses. We compared the distribution of potential confounding factors applying age-adjusted tests of trend across P quintiles. We estimated P levels across smoking categories applying ANOVA and post hoc (Tukey’s) tests. Initially, the association of P with mortality was assessed through Cox models, testing the proportionality assumption of the hazards via the Schoenfeld residuals test. All significant HRs from Cox’ models were found to be constant over follow-up time; therefore, we found no evidence for a time-dependent effect of P levels on mortality. In a second step, we compared the semi-parametric Cox model with parametric models, and found that Weibull regression models—albeit with highly similar results to Cox regressions—provide better statistical fit to the data than Cox models. Weibull models provided also better fit than the rest of parametric models. We applied Cox-Snell residuals graphs and Akaike (AIC) and Bayesian information criteria (BIC) to compare among models, as previously recommended [20]. Models with lower AIC and BIC correspond to a better fit. Therefore, the results reported in this manuscript correspond to Weibull regression models. Finally, we also performed competing-risks regressions models which allow for informative censoring due to the multiple possible causes of death [21]; these models provide an estimate of the effect of the exposure on the probability of developing the outcome over time [22].
Hazard ratios (HRs) are expressed per increase in 1 mg/dL (0.32 mmol/L) of P or in quintiles; the latter were built to explore a potential dose–effect relationship between phosphate levels and mortality.
The analysis time was set at the date of blood drawing. Subjects were followed until the first of the following events happened: death, lost to follow-up, or censoring by 1st January, 2012.
Adjustments were made firstly for age, BMI and smoking because they are related to mortality and P; subsequently other covariates that have been associated with mortality were added to the model and retained if they changed the beta estimate more than 10%, including eGFR, glucose, hsCRP, Mg levels, cholesterol to HDL cholesterol ratio, calcium, 25-hydroxyvitamin D and prevalent cardiovascular disease.
Results from RS-I and RS-II were meta-analyzed using fixed-effect model.
Primary analyses were done with subjects with complete information on covariates. Subsequently, missing values were imputed via multiple imputation with chained equations, allowing missingness at random. We followed specific guidelines for imputation for survival analysis.
Sensitivity analyses
We repeated analyses including only subjects with normal P (2.5–4.5 mg/dL; 0.81–1.45 mmol/L). We further adjusted the analyses for phosphate dietary intake and 1,25-dihydroxyvitamin D3 levels in a subset of participants from RS-I-1 (n = 4046).
Additionally, we performed stratified analyses according to smoking categories.
We used SPSS (version 21.0, Armonk, NY: IBM Corp), Stata (version 13, College Station TX: Stata Corp LP) and Comprehensive Meta-Analysis (version 2.2, Biostat, Englewood, NJ). A two-sided p <0.05 was considered significant.
Results
Serum phosphate correlates
A general descriptive summary of main continuous covariates is depicted in Table 1. The distribution of relevant covariates and risk factors across even quintiles of P for RS-I and RS-II is depicted in Table 2. P was higher in women than men in both cohorts (pdifference < 0.001). P levels were different across smoking categories in both sexes and cohorts (ANOVA p < 0.001); this difference was due to higher P in current smokers (Tukey’s tests > 0.05 between former and never smokers).
P was within normal range in 95.5 and 94.9% of participants in the fasting state (RS-I and RS-II, respectively) and in 89.7% of participants in the non-fasting state (RS-I-1).
Serum phosphate and all-cause mortality
During 14.5 year (median) and 10.9 year (mean) follow-up a total of 1631 and 469 fatal events occurred in RS-I and RS-II, respectively. We found a significant interaction between P and sex for all-cause mortality in RS-I (pinteraction < 0.001) and performed sex-stratified analyses. The results for the comparison of goodness-of-fit between parametric models and the semiparametric Cox model are displayed in Supplementary Table 1 (AIC and BIC criteria) and in Fig. 2 (Cox-Snell residuals plot). Both methods showed that Weibull models provide a better fit to our data among the parametric and semiparametric models.

Cox-Snell residuals plot for parametric models in the association between serum phosphate levels and all-cause mortality in men
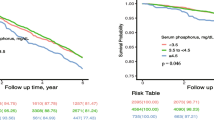
The associations between P and all-cause mortality are depicted in Table 3. Results from RS-I and RS-II were meta-analyzed (pooled HR (95% CI)). A significant association between P and all-cause mortality was found in men (1.46 (1.26–1.69)) but not in women (0.90 (0.77–1.05)). Adjustments in a full model composed of age, BMI, smoking, prevalent cardiovascular disease and levels of calcium, 25-hydroxyvitamin D, eGFR, CRP, Mg, glucose and total cholesterol to HDL cholesterol ratio levels did not substantially modify results (men: 1.49 (1.27–1.74); women: 0.92 (0.79–1.07)).
Similarly, results from RS-I-1 with non-fasting phosphate showed a significant association of phosphate with all-cause mortality in men (1.12 (1.02–1.23); no events = 1389), but not in women (0.99 (0.91–1.08); no events = 1779).
To explore whether there was a dose–response pattern in the association we found in men, we analyzed P in even quintiles and all-cause mortality in RS-I, the cohort with most events, (Table 4) and set the first quintile (lowest) as reference. We observed a significant trend for increasing P and mortality (ptrend < 0.001) with significant HRs for the fourth (1.35 (1.08–1.69)) and fifth quintile (1.49 (1.19–1.86)) compared with the first quintile.
Sensitivity analyses
Results after excluding subjects with abnormal P were similar to the unrestricted analyses (men: 1.44 (1.21–1.70); women: 0.87 (0.74–1.03)). Adjustments for phosphate and energy intake in men from RS-I-1 did not modify the results between non-fasting phosphate and all-cause mortality (1.13 (1.02–1.24); no events = 1117). Further adjustments for 1,25 dihydroxyvitamin D3 levels in a subset from RS-I-1 did not modify results (data not shown).
Serum phosphate and cause-specific mortality in men
We did not observe associations between P and cause-specific mortality in women (data not shown). In contrast, the pooled results in men (Table 5) showed a significant positive relation between P and CVD mortality (1.66 (1.29–2.14)). Exclusion of male subjects with prevalent CVD disease yielded similar results (1.69 (1.28–2.23)).
We also found an association between higher P and chronic lung disease mortality (1.94 (1.02–3.72)). Most of these cases clustered within COPD mortality. Therefore, we further investigated such a relation (Table 6), and found a significant association (4.44 (2.08–9.49)). Most likely due to power constraints, this association was not significant in RS-II (05 cases in contrast to 28 cases in RS-I) but there was no evidence for statistical difference between both estimates (pheterogeneity = 0.780). Further adjustments for glomerular filtration rate did not abolish the association between P and COPD mortality (4.16 (2.05–8.43)). Furthermore, the association was found to be consistent in subjects without chronic kidney disease (CKD) (6.58 (2.59–16.7)); whereas we found no association in subjects with CKD (1.14 (0.20–6.63)), although the latter analysis is constrained due to low number of events and driven only by RS-I. Non-fasting phosphate levels and COPD mortality in men from RS-I-1 also displayed a significant association (1.54 (1.05–2.27), no events = 69).
P was also found to be positively associated with mortality from other causes (1.67 (1.16–2.40)).
We found no significant associations between P and death due to cancer, infections, dementia or external causes.
Results from competing-risks regression models were similar to Weibull models and showed a significant association between P and mortality due to CVD (1.50 (1.12–2.02)), other causes (1.40 (1.01–1.93)) and COPD (2.42 (1.62–3.63)); no other significant associations were found (Supplementary Tables 2 and 3).
Analyses after applying multiple imputation yielded significant associations for P and all-cause, CVD, COPD and other causes of mortality in men (data not shown). Missingness of covariates of interest was less than 6%.
Sensitivity analyses
Results after excluding male subjects with abnormal P were similar to the unrestricted analyses (Supplementary Tables 4 and 5). Likewise, our findings remained essentially unaltered after adjustments for calcium and 25-hydroxyvitamin D levels; and were only slightly attenuated after further adjustments for levels of calcium, 25-hydroxyvitamin D and eGFR (CVD 1.65 (1.27–2.14), COPD 3.79 (1.87–7.69), other causes 1.76 (1.21–2.56)). Similar results were obtained after adjustments for cystatin-based eGFR. Additionally, the analyses after exclusion of male subjects with eGFR < 60 mL/min showed a positive association between P and mortality due to other causes (1.72 (1.13–2.61)) and COPD (6.58 (2.59–16.7)) - as previously mentioned- and a borderline association between P and CVD mortality (1.36 (1.00–1.85)).
Smoking adjustment did not attenuate the association between P and CVD or COPD mortality (data not shown). The results from the stratified analyses according to smoking categories (Supplementary Tables 6 and 7) showed that in studies combined the associations between P and all-cause and CVD mortality were in the same direction and did not show statistical evidence for a difference across categories (pheterogeneity = 0.752 for all-cause mortality and pheterogeneity = 0.796 for CVD mortality). The relation between P and COPD mortality in men from RS-I (RS-II excluded due to few events) was not statistically different among former and current smokers (pheterogeneity = 0.494).
As previously mentioned, analyses in men from RS-I-1 showed that non-fasting phosphate levels were also associated with chronic lung disease mortality and COPD mortality, and these associations were not abolished after further adjustments for phosphate and energy intake: chronic lung disease mortality: 1.79 (1.19–2.68); no events = 59; COPD mortality: 1.87 (1.20–2.91), no events = 49.
Discussion
This prospective population-based cohort study among elderly demonstrated that P was positively associated with all-cause mortality in men but not in women, supporting an effect modification by sex previously described [9]. When analyzing in detail cause-specific mortality in men, we found that this association was driven by mortality due to CVD, COPD and other causes. The association between increasing P and the composite endpoint of fatal and non-fatal CVD incidence in non-CKD population in sex-combined analyses has been reported before but is still scarce [7,8,9]. Our results provide evidence of an association between higher P—even within normal range—and death due to CVD in men. On the other hand, to the best of our knowledge the association we found with COPD mortality is novel. These results remained significant after adjustments for several potential confounders, were observed also after restricting the analyses to subjects with normal P and showed no heterogeneity between cohorts.
Several mechanisms have to be considered when analyzing P and mortality, including phosphate being a marker of another risk factor or through direct pathogenic pathways.
First, P levels are regulated by a complex interplay of factors that have been linked to mortality, such as 1,25-dihydroxyvitamin D3, PTH and FGF23. Low levels of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D3 have been found to be associated with increased mortality [23]. Nevertheless, the vitamin D adjustments did not modify our results.
PTH abnormalities have also been associated with mortality. Primary excess of PTH is associated with increased cardiovascular mortality [24], but in this context P tends to be low. Secondary elevations of PTH in impaired kidney function have been inconsistently associated with mortality. This compensatory mechanism in CKD is triggered when eGFR falls below 47 mL/min [25]. Although PTH levels were not available, the proportion of patients in our cohorts with eGFR below that threshold was considerably low (4% in RS-I and 2% in RS-II) suggesting that secondary hyperparathyroidism is unlikely to explain our findings. Nevertheless, PTH values seem to rise within normal range in the general population without CKD [26] at higher thresholds of decreasing eGFR (< 120 mL/min); whether increasing PTH values within normal range are associated with all-cause mortality in the long term is unclear [27].
Other important players in P homeostasis that might underlie its associations with mortality are the phosphatonins FGF23 and α-Klotho [3, 4]. FGF23 is synthesized mainly in osteocytes [5] and requires the presence of α-Klotho to bind to its receptor with high affinity and for signaling [28]. FGF23/α-Klotho axis decreases P through increased urinary phosphate excretion and both molecules are anti-ageing factors [5]. Primary causes of excess FGF23, such as in hereditary hypophosphatemic rickets, have been associated with cardiovascular calcification in cases of excessive phosphate treatment. Secondary FGF23 elevation occurs in CKD at earlier stages than PTH [3, 25] in response to P retention, and it has been linked to increased mortality [29, 30]. Similar to PTH, FGF23 elevations within normal range have been described at high thresholds of eGFR in population without CKD [26]; FGF23 levels have also been associated with mortality in this setting [31]. Nevertheless, FGF23 seems not to induce vascular calcification in most studies [4, 32, 33].
Recently, soluble klotho has been linked to increased mortality in CKD patients [34] although the lack of a validated assay for its measurement might be a concern for some [30].
Another potential confounder could be smoking. Similar to previous reports [7], P was found to be higher in current smokers. Although adjustments for smoking did not alter our analyses, due to heavy current and former smoking in men it is difficult to fully dissect its effects. Nevertheless, in studies combined the stratified analyses by smoking status showed that the associations between P and all-cause and CVD mortality appeared to be of the same direction and similar magnitude across smoking categories. The group of former smokers—who had similar P as never smokers—displayed the most statistically significant associations possibly due to larger number of subjects in this category. Specifically, P was related to COPD mortality comparably in current and former smokers men from RS-I but only significant in the latter group; a relation in non-smokers could not be tested due to low numbers in this subgroup. Therefore we do not anticipate that current smoking explains the association between P and COPD mortality.
Regarding direct effects, P itself is able to induce vascular calcification, a process with high resemblance to bone ossification and that increases mortality [33, 35]. Several pathways are known such as (a) differential gene expression in vascular smooth muscle cells with up-regulation of markers critical for mineralization [36]; and (b) elastin degradation, thought to be mediated by P induction of matrix metalloproteinase (MMP)-9.
The association we found between P and COPD mortality has never been described in humans before; interestingly there is additional evidence for the pathogenicity of high P stemming from rodent models with fgf23 or klotho knockout. These animals display similar phenotypes characterized by severe hyperphosphatemia and features of premature aging, such as osteoporosis, ectopic calcifications, pulmonary emphysema and short life span [37,38,39]. Heterozygous klotho mice also display emphysematous lungs. Remarkably, a low phosphate diet is able to alleviate or rescue the phenotype -including the lung emphysema; and a high phosphate diet worsens it [40], strongly suggesting that phosphate itself accelerates ageing [41] and induces alveoli destruction, and that this process can be modified by diet manipulation [40].
A new concept of phosphotoxicity as a risk factor for mammalian ageing has emerged lately [3, 40] and there are concerns that increasing phosphate intake through food additives may negatively influence multiple aspects of health [42]. Indeed it has been shown that high absolute P intake was positively related to all-cause mortality -not explained by CVD mortality [11]. Recently, a healthy diet—according to the Alternate Healthy Eating Index (2010) score—was associated with lower risk of COPD in humans [43]; interestingly in men but not women this beneficial association was driven mostly by a drastic reduction in red and processed meat consumption, expected to contain high phosphate [42]. A positive relation between cured meat intake and COPD risk has previously been reported in cross-sectional (NHANES III) and prospective studies [44, 45]. Importantly, when spirometric definitions for lung volumes and COPD have been applied, cured meat intake has been shown to be negatively associated with lung function, and positively related with COPD risk [44, 46]; the latter study showed that these associations were found predominantly in men. Cured meat consumption has also been shown prospectively to increase the hospital readmission rate in COPD patients [47].
From a mechanistic point of view, previous research [48] has shown that phosphate is able to directly induce injury in mice and human lung epithelial cells through increased DNA oxidative stress and apoptosis; indeed phosphate medium is used experimentally to induce oxidative lung injury. Interestingly, α-Klotho exerts protective antioxidant effects against lung injury induced by P [48], hyperoxia, and acute α-Klotho deficiency [49]. These data show that lung tissue is a target for phosphotoxic insult. Remarkably, increased P intake down-regulates α-Klotho expression in rodents [41]; therefore low P diet may be a therapeutic strategy to increase Klotho [3].
A genetic variant associated with low FGF23 was found to be associated with emphysema in smokers with COPD. More studies are needed to elucidate further the underlying mechanisms, especially considering that COPD ranks high in the most common causes of death worldwide.
The reasons for the sex difference between P and mortality are not clear. Interestingly, the vascular calcification induction by P is attenuated by 17β-estradiol, suggesting a potential hormonal reason for this difference [50]. Despite the fact that menopause is characterized by low estradiol levels, hormone replacement therapy-naïve postmenopausal women with higher 17β-estradiol levels display lower coronary calcification scores than those with lower 17β-estradiol [51]. Additionally, coronary infusion of 17β-estradiol exerts vasodilation in postmenopausal women, but not men [52]. Testosterone and estradiol play important roles as P regulators [19].
Although men had a less healthy profile at baseline than women, multiple adjustments did not abolish our results. Moreover, a previous study showed that P is associated with subclinical atherosclerosis in men (but not women) without prevalent cardiovascular and cerebrovascular disease at baseline [53].
This study has several limitations. 1,25-dihydroxyvitamin D3 levels were available only in a subgroup. PTH and FGF23 measurements were not available and it is known that kidney function in elderly can be misclassified even by eGFR. Our findings cannot be generalized to other ethnicities other than European Caucasians. Nevertheless, there are several strengths, such as the availability of two well-characterized cohorts with long follow-up, the detailed information on cause-specific mortality and the availability of multiple potential confounders. The completeness of follow-up was high (94 and 92% in RS-I and RS-II) indicating that obtained estimates are valid.
In conclusion, we found that higher P is associated with increased all-cause mortality and cause-specific mortality due to CVD, COPD and other causes in elderly men but not in women, adding more evidence for a modification of these associations by sex. We hereby provide evidence to support that the concept of phosphotoxicity also among non-CKD general population deserves further attention and, if causally related, it occurs independently of vitamin D levels and kidney function. Our study suggests that moderation of phosphate intake might be relevant also in non-CKD population for healthy ageing. Finally, we consider that the available evidence calls for a review of the currently accepted normal range of P. Further research is needed to clarify the underlying mechanisms, especially for COPD mortality, and to elucidate the reasons for the sex difference in the association of P with mortality.
References
Koeppen BM, Stanton BA. The renal system. In: Koeppen BM, Stanton BA editors. Berne & Levy physiology, 6th edn. Philadelphia, PA; 2010. p. 557-636.
Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–8.
Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–33.
Lim K, Lu TS, Molostvov G, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125(18):2243–55.
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131–55.
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31(4):607–17.
Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167(9):879–85.
Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G, Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation. 2005;112(17):2627–33.
Onufrak SJ, Bellasi A, Cardarelli F, et al. Investigation of gender heterogeneity in the associations of serum phosphorus with incident coronary artery disease and all-cause mortality. Am J Epidemiol. 2009;169(1):67–77.
Chang AR, Grams ME. Serum phosphorus and mortality in the Third National Health and Nutrition Examination Survey (NHANES III): effect modification by fasting. Am J Kidney Dis. 2014;64(4):567–73.
Chang AR, Lazo M, Appel LJ, Gutiérrez OM, Grams ME. High dietary phosphorus intake is associated with all-cause mortality: results from NHANES III. Am J Clin Nutr. 2014;99(2):320–7.
Ikram MA, Brusselle GGO, Murad SD, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol. 2017;32(9):807–50.
Bolland MJ, Grey AB, Ames RW, et al. The effects of seasonal variation of 25-hydroxyvitamin D and fat mass on a diagnosis of vitamin D sufficiency. Am J Clin Nutr. 2007;86(4):959–64.
Tomson J, Emberson J, Hill M, et al. Vitamin D and risk of death from vascular and non-vascular causes in the Whitehall Study and meta-analyses of 12000 deaths. Eur Heart J. 2013;34(18):1365–74.
Terzikhan N, Verhamme KM, Hofman A, Stricker BH, Brusselle GG, Lahousse L. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur J Epidemiol. 2016;31(8):785–92.
Stevens PE, Levin A, Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825–30.
Levey AS, Stevens LA, Schmid CH, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration), et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–12.
Campos-Obando N, Castano-Betancourt MC, Oei L, et al. Bone mineral density and chronic lung disease mortality: the Rotterdam Study. J Clin Endocrinol Metab. 2014;99(5):1834–42.
Meng J, Ohlsson C, Laughlin GA, Osteoporotic Fractures in Men (MrOs) Study Group, et al. Associations of estradiol and testosterone with serum phosphorus in older men: the Osteoporotic Fractures in Men study. Kidney Int. 2010;78(4):415–22.
Collet D. Modelling survival data in medical research. 3rd ed. London: Chapman & Hall/CRC; 2014.
Putter H, Fiocco M, Geskus RB. Tutorial in biostatistics: competing risks and multi-state models. Stat Med. 2007;26(11):2389–430.
Austin PC, Fine JP. Accounting for competing risks in randomized controlled trials: a review and recommendations for improvement. Stat Med. 2017;36(8):1203–9.
Lee DM, Vanderschueren D, Boonen S, European Male Ageing Study Group, et al. Association of 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D and parathyroid hormone with mortality among middle-aged and older European men. Age Ageing. 2014;43(4):528–35.
Nilsson IL, Yin L, Lundgren E, Rastad J, Ekbom A. Clinical presentation of primary hyperparathyroidism in Europe–nationwide cohort analysis on mortality from nonmalignant causes. J Bone Miner Res. 2002;17(Suppl 2):N68–74.
Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–8.
Dhayat NA, Ackermann D, Pruijm M, et al. Fibroblast growth factor 23 and markers of mineral metabolism in individuals with preserved renal function. Kidney Int. 2016;90(3):648–57.
Yang B, Lu C, Wu Q, Zhan J, Zhao H, Cao Y. Parathyroid hormone, cardiovascular and all-cause mortality: a meta-analysis. Clin Chim Acta. 2016;455:154–60.
Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–4.
Yang H, Luo H, Tang X, et al. Prognostic value of FGF23 among patients with end-stage renal disease: a systematic review and meta-analysis. Biomark Med. 2016;10(5):547–56.
Munoz Mendoza J, Isakova T, Cai X, CRIC Study Investigators, et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017;91(3):711–9.
Qin Z, Liu X, Song M, et al. Fibroblast growth factor 23 as a predictor of cardiovascular and all-cause mortality in prospective studies. Atherosclerosis. 2017;261:1–11.
Scialla JJ, Lau WL, Reilly MP, Chronic Renal Insufficiency Cohort Study Investigators, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83:1159–68.
Yamada S, Giacelli CM. Vascular calcification in CKD-MBD: roles for phosphate, FGF23 and Klotho. Bone. 2017;100:87–93.
Marçais C, Maucort-Boulch D, Drai J, ARNOGENE project, et al. Circulating klotho associates with cardiovascular morbidity and mortality during hemodialysis. J Clin Endocrinol Metab. 2017;102(9):3154–61.
Wu M, Rementer C, Giachelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int. 2013;93(4):365–73.
Román-García P, Carrillo-López N, Fernández-Martín JL, Naves-Díaz M, Ruiz-Torres MP, Cannata-Andía JB. High phosphorus diet induces vascular calcification, a related decrease in bone mass and changes in the aortic gene expression. Bone. 2010;46(1):121–8.
Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51.
Nakatani T, Sarraj B, Ohnishi M, et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J. 2009;23(2):433–41.
Suga T, Kurabayashi M, Sando Y, et al. Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol. 2000;22(1):26–33.
Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24(9):3562–71.
Morishita K, Shirai A, Kubota M, et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr. 2001;131(12):3182–8.
European Food Safety Authority. Assessment of one published review on health risks associated with phosphate additives in food. EFSA Journal. 2013;11:3444. https://doi.org/10.2903/j.efsa.2013.3444.
Varraso R, Chiuve SE, Fung TT, et al. Alternate Healthy Eating Index 2010 and risk of chronic obstructive pulmonary disease among US women and men: prospective study. BMJ. 2015;350:h286.
Jiang R, Paik DC, Hankinson JL, Barr RG. Cured meat consumption, lung function and chronic obstructive pulmonary disease among United States adults. Am J Respir Crit Care Med. 2007;175(8):798–804.
Varraso R, Jiang R, Barr RG, Willet WC, Camargo CA Jr. Prospective study of cured meats consumption and risk of chronic obstructive pulmonary disease in men. Am J Epidemiol. 2007;166(12):1438–45.
Okubo H, Shaheen SO, Ntani G, Hertfordshire Cohort Study Group, et al. Processed meat consumption and lung function: modification by antioxidants and smoking. Eur Respir J. 2014;43(4):972–82.
de Battle J, Mendez M, Romieu I, PAC-COPD Study Group, et al. Cured meat consumption increases risk of readmission in COPD patients. Eur Respir J. 2012;40(3):555–60.
Ravikumar P, Ye J, Zhang J, et al. α-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol. 2014;307(7):L566–75.
Ravikumar P, Li L, Ye J, et al. αKlotho deficiency in acute kidney injury contributes to lung damage. J Appl Physiol. 2016;120(7):723–32.
Rzewuska-Lech E, Jayachandran M, Fitzpatrick LA, Miller VM. Differential effects of 17beta-estradiol and raloxifene on VSMC phenotype and expression of osteoblast-associated proteins. Am J Physiol Endocrinol Metab. 2005;289(1):E105–12.
Jeon GH, Kim SH, Yun SC, Chae HD, Kim CH, Kang BM. Association between serum estradiol levels and coronary artery calcification in postmenopausal women. Menopause. 2010;17(5):902–7.
Collins P, Rosano GM, Sarrel PM, et al. 17 beta-Estradiol attenuates acetylcholine-induced coronary arterial constriction in women but not men with coronary heart disease. Circulation. 1995;92(1):24–30.
Onufrak SJ, Bellasi A, Shaw LJ, et al. Phosphorus levels are associated with subclinical atherosclerosis in the general population. Atherosclerosis. 2008;199(2):424–31.
Acknowledgments
The authors thank the participants and staff of the research center of the Rotterdam Study. The authors also acknowledge Dr. F. Rivadeneira, for his critical review of the manuscript. The Rotterdam Study has been approved by the Medical Ethics Committee of the Erasmus MC (Registration Number MEC 02.1015) and by the Dutch Ministry of Health, Welfare and Sport (Population Screening Act WBO, License Number 1071272-159521-PG). The Rotterdam Study has been entered into the Netherlands National Trial Register (NTR; www.trialregister.nl) and into the WHO International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/network/primary/en/) under shared catalogue number NTR6831. All participants provided written informed consent to participate in the study and to have their information obtained from treating physicians.
Funding
The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The funding sources had no influence in the study design, collection, analysis, interpretation of data, writing of the report and in the decision to submit the article.
Author information
Authors and Affiliations
Contributions
Dr. Zillikens and N. Campos-Obando are the study guarantors and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: N. Campos-Obando and Dr. Zillikens. Acquisition of data: Prof. Hofman, Dr. Zillikens, Prof. Uitterlinden, Prof. Stricker, Prof. Brusselle, Prof. Franco, Dr. Lahousse. Analysis and interpretation of data: N. Campos-Obando and Dr. Zillikens. Drafting of the manuscript: N. Campos-Obando and Dr. Zillikens. Critical review of the manuscript for important intellectual content: all authors. Statistical analyses: N. Campos-Obando. Obtained funding: Prof. Hofman, Prof. Uitterlinden. Administrative, technical and material support: Dr. Zillikens, Prof. Uitterlinden. Study supervision: Dr. Zillikens.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Ethical approval
The Rotterdam Study was approved by the Medical Ethics Committee of Erasmus Medical Center.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Campos-Obando, N., Lahousse, L., Brusselle, G. et al. Serum phosphate levels are related to all-cause, cardiovascular and COPD mortality in men. Eur J Epidemiol 33, 859–871 (2018). https://doi.org/10.1007/s10654-018-0407-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-018-0407-7