
Summary
Background To determine the recommended phase II dose (RP2D) and assess the safety, pharmacokinetics (PKs) and pharmacodynamics of RO4929097in combination with temsirolimus. Methods Escalating doses of RO4929097 and temsirolimus were administered at three dose levels. Patients received once daily oral RO4929097 on a 3 days on/4 days off schedule every week, and weekly intravenous temsirolimus. Blood samples were collected for PK analysis. Archival tissue specimens were collected for Notch pathway biomarker analysis and genotyping of frequent oncogenic mutations. Results Seventeen patients with refractory advanced solid tumors were enrolled in three dose levels (DLs): DL1 (RO4929097 10 mg; Temsirolimus 25 mg), DL2 (RO4929097 20 mg; Temsirolimus 25 mg), and DL3 (RO4929097 20 mg; Temsirolimus 37.5 mg). The most common toxicities related to the study drug combination included: fatigue (82 %; grade 3 6 %), mucositis, (71 %; grade 3 6 %), neutropenia (59 %; grade 3 12 %), anemia (59 %; grade 3 0 %), and hypertriglyceridemia (59 %; grade 3 0 %). Two dose-limiting toxicities, grade 3 rash and grade 3 mucositis, were observed in the same patient in the first dose level prompting dose expansion. Eleven patients (73 %) had stable disease as their best response. Co-administration of RO4929097 was associated with increased clearance and reduced exposure to temsirolimus, suggestive of drug-drug interaction via CYP3A4 induction. No correlation between the expression of Notch pathway biomarkers or genotype and time to progression was noted. Conclusions RO4929097 can be safely combined with temsirolimus in patients with advanced solid tumors. The RP2D was established at 20 mg of RO4929097 combined with 37.5 mg of temsirolimus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Notch signaling pathway is highly conserved and plays a critical role in regulating cell-to-cell communication during embryogenesis, cellular proliferation, differentiation, and apoptosis [2]. Deregulation of Notch signaling has also been involved in several malignancies [8, 10]. Increased Notch signaling promotes tumor cell proliferation by maintaining tumor cells in a stem-cell–like proliferative state. As a result, Notch signaling inhibitors are being actively investigated for the treatment of cancer. Mammalian-membrane-bound Notch ligands consist of two structurally distinct families: Delta-like ligands 1, 3 and 4, and Jagged ligands 1 and 2 that interact with four transmembrane Notch receptors (Notch 1–4). Once ligand-receptor binding occurs, the Notch receptor undergoes a conformational change to expose previously protected sites to proteolytic cleavage by metalloprotease and γ-secretase, releasing the Notch intracellular domain (NICD) fragment [11]. NICD translocates to the nucleus where it forms a transcription-activating complex, thereby activating transcription of target genes. Blocking Notch signaling via inhibition of γ-secretase is an attractive therapeutic strategy. RO4929097 is a potent and selective inhibitor of γ-secretase that produces a less transformed, flattened, slower growing phenotype in a variety of cancer cell lines [7]. In vivo studies established antitumor activity in xenograft mouse models of colon, pancreatic, and non–small cell lung cancer with both continuous and intermittent dosing, which persisted after cessation of treatment [7].
A recently reported first-in-human phase I study evaluated two different oral dosing schedules for RO4929097 given as a single-agent [15]. RO4929097 was well tolerated at doses up to 135 mg when administered for 7 consecutive days followed by 14 days off treatment during a 21-day cycle. Administration of RO4929097 was also evaluated on a three days-on/four days-off schedule. No DLTs were observed in either dosing regimen. Pharmacokinetic (PK) analysis showed that RO4929097 exposure decreased after repeated dosing, due to reversible CYP3A4 autoinduction at doses as low as 24 mg in the 3-days on/4-days off schedule.
There is cross talk between the Notch and mammalian target of rapamycin (mTOR) signaling pathways. The Notch pathway regulates activation of both phosphatase and tensine homologue (PTEN) and PI3K/AKT signaling components in normal cells; however, the aberrant activation of Notch signaling pathway induces the direct stimulation of PI3K/mTOR pathway leading to tumor cell growth [8]. In vitro studies in pancreatic cancer cell lines have shown a synergistic antitumor effect mediated through enhanced AKT suppression when rapamycin and a γ-secretase inhibitor were administrated [16]. A recent in vitro study in T-cell acute lymphoblastic leukemia model has shown that PI3K/mTOR inhibition leads to upregulation of Notch-myc pathway. However, dual blockade of both PI3K/mTOR and Notch produces enhanced cell-cycle arrest and cell death, providing a rationale for evaluating temsirolimus in combination with RO4929097 [13].
The main objectives of this phase I clinical trial were i) to determine the recommended phase II dose (RP2D) and safety profile of temsirolimus in combination with RO4929097; and ii) to evaluate the PK and pharmacodynamic effects, and preliminary evidence of anticancer activity of the combination.
Patients and methods
Patient eligibility
Patients were required to have a histologically confirmed advanced, incurable solid malignancy that was refractory to conventional therapy or for which no standard therapy existed, age ≥18 years, life expectancy ≥12 weeks, an Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) ≤1, with adequate hematological, hepatic, and renal function. Prior therapy with a γ-secretase inhibitor, or any inhibitor of the PI3K/AKT/mTOR pathway was prohibited. Key exclusion criteria included: a) concomitant use of medications that were strong inducers/inhibitors or substrates of CYP3A4; b) patients with malabsorption syndrome or other condition that would interfere with intestinal absorption; c) uncontrolled hypocalcemia, hypomagnesemia, hyponatremia, hypophosphatemia or hypokalemia despite adequate electrolyte supplementation; d) a QTc interval ≥470 mSec as measured by Bazett’s formula; e) pre-existing significant pulmonary infiltrates of unknown origin.
The study was conducted at two institutions, the Princess Margaret Cancer Centre and the Juravinski Cancer Centre. The trial was approved by the Ontario Cancer Research Ethics Board. The study was registered at clinicaltrials.gov (NCT 01198184), sponsored by the Princess Margaret Hospital Phase I Consortium, and supported by the National Cancer Institute (NCI) Contract No. U01-CA132123.
Study design
This was an open-label, dose-escalation phase I trial. RO4929097 was orally administered on an empty stomach on a three days-on four days-off schedule, weekly; and temsirolimus was given over 30 min intravenously (i.v.) every week (Fig. 1). When both drugs were to be administered together on a given day, RO4929097 was administered first. Premedication with diphenhydramine 25–50 mg IV was given approximately 30 min before temsirolimus infusion to prevent potential hypersensitivity reactions. Cycles were every 21 days except for cycle 1, which was 28 days. Cycle 1 consisted of a run-in period of 1 week with temsirolimus given alone on day 1, allowing for PK evaluation of temsirolimus. For cycle 1 only, temsirolimus was given on days 1, 8, 15 and 22, while RO4929097 was given starting at cycle 1 day 8, using the 3/4 schedule (i.e. 3-days on/4-days off). From cycle 2 onwards, cycles were 21 days in duration with RO4929097 administered on days 1–3, 8–10, and 15–17 with weekly doses of IV temsirolimus on days 1, 8 and 15. Three dose levels were planned. Due to the observed auto-induction of RO4929097 metabolism at doses above 20 mg in the monotherapy trial [15], dose escalation of R04929097 above 20 mg was not planned. DL3 included 37.5 mg temsirolimus in order to achieve blood levels equivalent to the standard 25 mg weekly dose in the event of a potential drug-drug interaction with RO49092907 mediated by CYP3A4.

Treatment schedule. Cycle 1 was 28-day long. Cycle 1, day 1 started with single-agent temsirolimus administration, allowing for pharmacokinetic sampling, RO4929097 was first administered at day 8. From cycle 2 onwards, cycle duration was 21 days. Temsirolimus was administered weekly and RO4929097 was given on a 3-days on/4-days off schedule, prior to temsirolimus administration
Toxicity was graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events version (NCI CTCAE v4.0). Dose-limiting toxicities (DLTs) were defined as adverse events (AEs) as least possibly related to study medication(s) and fulfilling one of the following criteria: a) grade 3 or 4 neutropenia complicated by fever ≥38.5 °C, or grade 4 neutropenia of at least 7 days duration; b) grade 3 thrombocytopenia complicated by hemorrhage or grade 4 thrombocytopenia; or c) any grade 3 or higher non-hematologic toxicity (except anorexia, nausea, vomiting, and diarrhea that was not optimally controlled with appropriate medical intervention); d) any ≥grade 3 electrolyte abnormality including hypophosphatemia, hypocalcemia, or hypomagnesemia that did not resolve within 72 h with appropriate therapy or if it was associated with new ECG changes; e) treatment-related toxicities that resulted in failure to receive at least 75 % of the planned doses of RO4929097 and of temsirolimus despite maximal supportive care measures; f) inability to resume dosing for cycle 2 at the current dose level within 14 days due to treatment-related toxicity.
A standard 3 + 3 design was used. Dose cohorts initially included 3 patients. If 1 patient out of 3 experienced a DLT, 3 additional patients were treated at that dose level. If only 1 out of 6 patients in that dose level experienced a DLT, escalation to the next level occurred. If 2 out of 3 or 2 out of 6 patients experienced DLT, no further dose escalation occurred. The Recommended Phase II Dose (RP2D) (equivalent to Maximum Tolerated Dose (MTD)) was defined as the dose level at which ≤1/6 patients experienced DLT. No intra-patient escalation was allowed. Patients could continue on study treatment until progressive disease was documented. Low dose, non-myelosuppressive radiotherapy for symptomatic palliation was permitted.
Patients who did not complete cycle 1 due to reasons besides DLT (e.g. early withdrawal of consent, intercurrent illness unrelated to study drugs, or symptomatic disease progression) were replaced and were not considered evaluable for DLT.
Patient evaluation
Pre-treatment evaluations were performed within 2 weeks of treatment initiation and included history and physical examination, ECOG PS, hematology, serum chemistry, urinalysis, and electrocardiogram (ECG). Physical examinations, hematology and chemistry were repeated weekly during the first cycle; and on day 1 of each subsequent cycle from cycle 2 onwards. Baseline radiological investigations were performed within 28 days of treatment initiation. Objective tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 every two cycles [5].
Dose modifications
Patients were required to meet the following criteria to receive study drugs on day 1 of each treatment cycle: absolute neutrophil count ≥1.5 × 109, platelets ≥75 × 109, normal serum electrolytes, non-hematologic toxicity recovered to grade ≤1, and no evidence of disease progression.
Patients were withdrawn from the study if they failed to recover to grade ≤1 or tolerable grade 2 (or within 1 grade of starting values for pre-existing laboratory abnormalities) from a treatment-related toxicity within 14 days or if they experienced an agent-related AE that required dose modification despite the number of permitted dose reductions (Supplementary Table 1). If one agent was discontinued due to toxicity, patients could continue to receive the other agent if, in the opinion of the treating physician, the patient was benefitting from treatment.
Study treatment continued until disease progression, an unacceptable AE, withdrawal of consent, or changes in the patient’s condition rendering further treatment unacceptable.
Pharmacokinetic (PK) analysis
Blood samples for RO4929097 and temsirolimus were collected serially for PK analysis during cycle 1 and 2 (Supplementary Tables 2 and 3). The unbound RO4929097 fractionswere obtained by filtrating plasma samples using AmicoCentrifree® Micropartition devices (Millipore Corp., Bedford, MA, USA). Plasma temsirolimus, total and unbound RO4929097 concentrations were determined using validated HPLC-tandem mass spectrometry methods [9, 17]. Pharmacokinetic parameters were calculated by non-parametric methods using WinNonlin (Version 5.3, Pharsight Corp., Sunnyvale, CA).
Notch pathway biomarkers
Archival paraffin-embedded tumorspecimens wererequested on all subjects for immunohistochemical analysis of components of the Notch pathway: Jagged-1, (NICD) and Notch-3. Immunohistochemistry was performed using standard techniques. Briefly, 4 μm paraffin-embedded tumor sections were preheated to 57° for 15 min, cooled to room temperature, dewaxed in xylene and rehydrated in graded alcohols. Endogenous peroxidase activity was blocked in 0.3 % hydrogen peroxide in PBS for 30 min. Heat induced epitope retrieval was accomplished using 10 mM citrate buffer (pH 6) in Biocare Digital DecloakingChamber (using factory recommended settings for IHC). Anti-NICD (Cell Signaling #2421; 1/50) and anti-Notch-3 (Santa Cruz Biotechnologies #sc-5593; 1/150) staining was performed using VECTASTAIN ABC Kit (Rabbit IgG) (Vector Labs #PK-4001) with the following modifications: permeablization with 0.3 % Triton X100 in PBS for 2 × 10 min before block; block for 1 h room temperature in PBS 3 % BSA, 20 mM MgCl2, 5 % FBS, 0.3 % Tween20, plus Vector blocking sera); incubation with primary antibody (diluted in blocking buffer plus vector blocking sera) overnight at 4°; secondary antibody diluted 1/100 in PBS 5 % BSA plus Vector blocking sera; all washes 2 × 5 min PBS, followed by 2 × 5 min PBS 0.3 % Triton X100. Anti Jagged-1 (R&D Systems #AF1277; 1/50) staining was performed using Cell and Tissue Staining kit (Goat IgG) (R&D Systems #CTS008) with a 4° overnight incubation with primary antibody. All immunohistochemistry was performed using Shandon Sequenza immunostaining coverplates (Fisher #7219950) and Peroxidase Substrate Kit, DAB (Vector Labs #SK-4100). Slides were counterstained with hematoxylin, dehydrated in graded alcohols to xylene, and mounted using Permount.
The stained slides were scored centrally (the pathologist, B.A.C., was blinded to the clinical outcome) for the presence of Jagged-1, NICD, and Notch-3. The positive antibody reaction was scored into four grades, according to the intensity of the staining: 0, 1+, 2+ and 3+. The percentage of positive cells was also scored into four categories: 0 (0 %), 1 (1–33 %), 2 (34–66 %), and 3 (67–100 %). The product of the intensity and the percentage scores was used as the final score [1, 3].
Mutational analysis
In patients with available archival paraffin-embedded tumor specimens and sufficient DNA, genotyping was performed with either Sequenom MassARRAY (Sequenom) PMH v1.0 customized panel for solid tumors that includes 280 mutations in 23 genes or the MiSeq (Illumina) TruSeq Amplicon Cancer Panel (TSACP) panel that includes 212 amplicons in 48 genes in the CLIA-certified University Health Network Advanced Molecular Diagnostics Laboratory.
Statistical analysis
Standard descriptive statistics, such as the mean, median, range and proportion, were used to summarize the patient sample and frequency of AEs. Wilcoxon test was performed for comparison of Notch biomarker protein expression between patient subgroups. A fixed-effect ANOVA test was used to compare temsirolimus PK parameters at three different time points during cycle 1 (day 1, day 8 and day 15). Time to progression (TTP) was defined as the time from start of treatment until disease progression. Radiologic assessment of disease status was repeated every 6 weeks. Univariate Cox proportional hazard analysis was performed to assess the association between protein expression and TTP. Exact 95 % confidence intervals were calculated for each proportion of interest. Statistical analysis was performed using SAS 9.2 software.
Results
Patient demographics
Between August 2010 and March 2012, 17 patients were enrolled (7 males; 10 females). Median age was 62 (range 28–84); most patients were ECOG PS 1 and had been treated with a median of 3 lines of chemotherapy (Table 1). Most common tumor types were sarcoma (n = 6, 35 %), neuroendocrine (n = 2, 12 %) and squamous cell carcinoma of the head and neck (n = 2, 12 %).
Dose escalation and RP2D
Eight, three, and six patients were enrolled in dose levels (DL) 1, 2, and 3, respectively. Treatment is summarized in Table 2. Two patients in DL1 (RO4929097 10 mg, Temsirolimus 25 mg) were non-evaluable for DLT. One patient voluntarily withdrew consent at day 22. Another non-compliant patient in the first cohort did not receive two of the four planned doses of temsirolimus. The two missed doses could not be attributed to toxicity. Thus, this patient was considered not evaluable for DLT. One patient in DL1 experienced two DLT events (grade 3 oral mucositis and grade 3 maculo-papular rash), prompting treatment of three additional patients at this dose level. No additional DLT events were observed in the expanded DL1 allowing for further dose escalation to DL2 (n = 3). In DL2 and DL3, 0/3 patients initially treated experienced DLT. An additional 3 patients were treated at DL3 to better evaluate safety of this dose level. No DLTs were observed (0/6 patients treated in DL3), leading to a declaration of 20 mg of oral RO4929097 in a 3-days-on/4-days-off schedule along with 37.5 mg of temsirolimus as the RP2D.
Safety and compliance
All 17 treated patients were evaluable for toxicity. The most frequently reported treatment-related AEs are listed in Table 3.
Fatigue (82 %) and oral mucositis (71 %) were the most frequently reported non-hematological treatment-related AEs. These were grade 1 and 2 in most cases and were easily managed with standard supportive measures. Only one episode of grade 3 mucositis was observed. Maculo-papular rash was observed in 7 patients (41 %), with only one grade 3 episode. Three patients (2 patients with grade 2; 1 patient with grade 3) required treatment interruption due to rash. Rash was reversible in all cases and no dose modification was needed when treatment was resumed. Gastrointestinal toxicities, including anorexia (47 %), nausea (41 %), vomiting (29 %), and diarrhea (18 %) were also frequently observed. Consistent with the single agent safety profile of RO4929097 and temsirolimus, hypophosphatemia was frequently observed (47 %). However, most episodes were grade 2, and were managed with oral and/or intravenous supplementation. Prolongation of the QTc interval has been reported with RO4929097. In this study, three patients (17 %) experienced grade 1 asymptomatic transient QT interval prolongation. This was not associated with electrolyte abnormalities. No clinically significant rhythm abnormalities were noted.
Metabolic disturbances and pneumonitis have been frequently described with temsirolimus [12]. In this study, hypertriglyceridemia was the most commonly reported (n = 10, 59 %) metabolic AE. Most cases were grade 1 and 2 in severity, and no intervention was required. A few instances of hyperglycemia (n = 4, 23 %) and hypercholesterolemia (n = 2, 12 %) were noted. Hyperglycemia was of grade 1 or 2 severity in all cases, and was managed with diet and/or oral antidiabetic medication. One patient with prior history of hypercholesterolemia required statin treatment for grade 3 hypercholesterolemia. Another patient received low-dose rosuvastatin for grade 2 hypercholesterolemia while on treatment and maintained an adequate control throughout the study duration. No dose modifications or study treatment interruption was required for any metabolic abnormalities. No episodes of pneumonitis were observed in this study.
Neutropenia (59 %) and anemia (59 %) were the most common hematological toxicities. There were two grade 3 episodes of neutropenia that were not associated with infection. No patient discontinued study treatment due to hematological toxicity.
Dose delays and dose modifications for RO4929097 and temsirolimus are provided in Supplementary Table 4.
Antitumor activity
No objective responses were observed. Eleven patients (73 %) had stable disease as their best response and six patients (40 %) completed at least six cycles of treatment (4 months) without experiencing disease progression. The median progression-free survival was 4.2 months (95 % confidence interval [CI], 1.5–8.7 months). Two patients, diagnosed with a metastatic high-grade synovial sarcoma and a metastatic gastrointestinal stromal tumor respectively, who had disease progression to their last regimen, remain on study treatment at cycle 13 and 17 at the time of data cutoff (Nov 5, 2012).
Pharmacokinetics
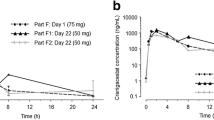
Plasma exposure of RO4929097 was measured at day 8 and day 22 (Fig. 2). A dose-proportional increase in RO4929097 exposure was not observed. Temsirolimus clearance was significantly increased from cycle 1 day 1 to cycle 1 day 15 across all doses levels (p < 0.01), but not between cycle 1 days 1 and 8 (Fig. 3a). Correspondingly, there was a significant reduction in temsirolimus area under the curve (AUC) from cycle 1 day 1 to cycle 1 day 15 (p < 0.01),but not between cycle 1 days 1 and 8 (Fig. 3b). No difference in peak temsirolimus plasma concentrations (Cmax) was observed among different dosing days in cycle 1. These findings are consistent with a cumulative effect of repetitive RO4929097 dosing on CYP3A4 induction leading to increased temsirolimus clearance and reduced AUC.

Plasma exposure of RO4929097 (area under the curve [AUC]) at day 8 and day 22, at the different dose levels. Horizontal bars represent standard deviation. Dose level 1: RO4929097, 10 mg; temsirolimus 25 mg. Dose level 2: RO4929097, 20 mg; temsirolimus 25 mg. RO4929097, 20 mg; temsirolimus 37.5 mg

a Temsirolimus clearance (Cl) observed at cycle day 1, day 8 and day 15 for the different dose levels. Horizontal bars represent standard deviation. Dose level 1: RO4929097, 10 mg; temsirolimus 25 mg. Dose level 2: RO4929097, 20 mg; temsirolimus 25 mg. RO4929097, 20 mg; temsirolimus 37.5 mg. (*) denotes p value for comparison between cycle 1 day 1 and cycle 1 day 8; (**) denotes p value for comparison between cycle 1 day 1 and cycle 1 day 15. b Temsirolimus area under the curve (AUC) observed at cycle 1, day1, day 8 and day 15 for the different dose levels. Horizontal bars represent standard deviation. Dose level 1: RO4929097, 10 mg; temsirolimus 25 mg. Dose level 2: RO4929097, 20 mg; temsirolimus 25 mg. RO4929097, 20 mg; temsirolimus 37.5 mg. (*) denotes p value for comparison between cycle 1 day 1 and cycle 1 day 8; (**) denotes p value for comparison between cycle 1 day 1 and cycle 1 day 15
Notch pathway biomarkers
Archival pathology specimens were available from 14 patients. Immunohistochemical (IHC) expression of Notch receptor ligand Jagged-1, Notch-3 receptor, and Notch intracellular domain (NCID) was performed on paraffin-embedded tumor tissue. The median Jagged-1 IHC intensity score was 6 (range 3–7). The median Notch-3 score was 2 (range 0–7), and the median NCID score was 5 (range 0–8). No significant differences in the median protein expression score were seen between patients who remained progression-free after four cycles of treatment and those who progressed prior to cycle 5. No significant association was detected between protein expression of any of the Notch biomarkers evaluated and time to progression (Supplementary Table 5). Of 14 tumors with archived pathology specimens and sufficient DNA available for further analysis, mutations were identified in 5 patients (Supplementary Table 6). No correlation between mutational status (mutation(s) vs no mutation(s)) and TTP was found.
Discussion
This study evaluated the feasibility of the combination of RO4929097, an oral γ-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. The RP2D of such combination was determined to be 20 mg of RO4929097 in 3-days on/4-days off weekly schedule along with the weekly administration of 37.5 mg of temsirolimus. The most common treatment-related toxicities of the combination were fatigue and mucositis. Other toxicities commonly seen with temsirolimus were not exacerbated by the addition of RO4929097 [4, 12].
It has been observed that RO4929097 exposure decreases after repeated oral administration due to reversible CYP3A4 auto-induction at doses as low as 24 mg in the 3-days on/4-days off schedule [15]. Thus, it was decided to start RO4929097 dosing at 10 mg, and RO4929097 dose was only escalated to 20 mg. However, even at these relatively low doses, RO4929097 appears to induce CYP3A4 activity, resulting in a significant increase in temsirolimus clearance and a significant decrease in temsirolimus exposure.
No responses were seen with the combination of RO4929097 and temsirolimus. Six evaluable patients (40 %) received at least six cycles of treatment, with SD as best response. Of note, two patients, one with a gastrointestinal stromal tumor and the other with a high-grade synovial sarcoma, remain on treatment after more than 12 cycles.
This study has several limitations. Based on the previously observed auto- induction phenomenon with doses of RO4929097 above 24 mg, RO4929097 was given at low doses (10 mg in DL1; 20 mg in DL2 and DL3). However, human doses of R04929097 ≥ 6 mg achieved plasma concentrations that exceed efficacious exposure levels established in animal models (exposures producing efficacy (AUC24h) were approximately 1,100 ng h/mL after oral daily dosing schedule of 10 mg/kg/day for 21 days in rats, equivalent to 9 mg/day in humans). In fact, the lowest dose level of RO4929097 tested in this study (10 mg) produced serum concentrations above the minimum effective concentration required to inhibit gamma secretase in xenograft studies (data not shown). Secretory diarrhea is a potential pharmacodynamic marker of Notch inhibition due to the critical role of Notch in proper cell differentiation of the rapidly self-renewing crypt cells of the small intestine [7]. Inhibiting γ-secretase shifts the balance of celltypes from nutrient-absorbing cells tomucus-secreting cells leading to secretory diarrhea. Diarrhea was infrequently observed in our study (3 patients, 18 %, all grade 1 episodes), suggesting that the doses and schedule of RO4929097 tested may not have been adequate to inhibit Notch. Other putative pharmacodynamic markers, including circulating Aß-40 protein and VEGFR2 protein levels in plasma, changes in interleukin-6 plasma levels [15], andexpression of Notch pathway transcripts in hair follicles, have been evaluated in studies of γ-secretase inhibitors, although it is not clear if these biomarkers reflect intratumoral Notch inhibition [15]. However, this study did not include such pharmacodynamic assessments. Future studies may require pre- and post-treatment paired tumor biopsies in order to assess target engagement and the pharmacodynamic effects of combined pathway inhibition.
Exploratory analysis of selected components of the Notch signaling pathway (Jagged-1, NICD, and Notch-3 receptor) evaluated by immunohistochemistry was performed using paraffin embedded archival tumor specimens. There was no association between protein expression of any of the Notch components and progression free survival beyond 4 cycles, although the number of patients included in this analysis was limited. Most patients had been previously treated with several chemotherapy regimens prior to starting on study treatment. It is unknown whether exposure to prior cytotoxic treatment affects the expression of different Notch pathway components. Future studies should investigate whether expression of Notch pathway components in tumor biopsies acquired before starting treatment is associated with clinical outcome with Notch inhibitor therapy. Our study performed targeted genotyping of tumors with sufficient DNA for analysis. We did not find an association between the mutational status and clinical outcome. This may be due to small numbers and the mixed tumor types of the patients tested.
Notch signaling is an area of intense research for the development of new anticancer therapies. Beyond γ-secretase inhibitors, other approaches currently being investigated include therapeutic antibody targeting of individual notch receptors and their Dll4 ligand [6, 14, 18]. The available clinical data from studies of γ-secretase inhibitors suggest limited activity in advanced solid tumors as monotherapy. The future of Notch pathway inhibition may be in combination with cytotoxic and other targeted therapies. Our study demonstrates that RO4929097 and temsirolimus can be safely combined. Our trial was prematurely discontinued before planned expansion cohorts at the RP2D in endometrial and renal cell cancers could be evaluated because the clinical development of RO4929097 was stopped due to its unfavorable pharmacological profile. Future studies are warranted to evaluate if combined Notch and PI3K/mTOR inhibition is an active anti-cancer approach.
References
Allred DC, Harvey JM, Berardo M, Clark GM (1998) Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 11:155–168
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776
Dickson BC, Mulligan AM, Zhang H, Lockwood G, O’Malley FP, Egan SE, Reedijk M (2007) High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol 20:685–693
Duran I, Siu LL, Oza AM, Chung TB, Sturgeon J, Townsley CA, Pond GR, Seymour L, Niroumand M (2006) Characterisation of the lung toxicity of the cell cycle inhibitor temsirolimus. Eur J Cancer 42:1875–1880
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Jenkins DW, Ross S, Veldman-Jones M, Foltz IN, Clavette BC, Manchulenko K, Eberlein C, Kendrew J, Petteruti P, Cho S, Damschroder M, Peng L, Baker D, Smith NR, Weir HM, Blakey DC, Bedian V, Barry ST (2012) MEDI0639: a novel therapeutic antibody targeting Dll4 modulates endothelial cell function and angiogenesis in vivo. Mol Cancer Ther 11:1650–1660
Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, Roberts J, Cai J, Berkofsky-Fessler W, Hilton H, Linn M, Flohr A, Jakob-Rotne R, Jacobsen H, Glenn K, Heimbrook D, Boylan JF (2009) Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 69:7672–7680
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zuniga-Pflucker JC, Dominguez M, Ferrando AA (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13:1203–1210
Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C, Korth-Bradley J, Hanauske A, Armand JP (2004) Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol 22:2336–2347
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE (2005) High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 65:8530–8537
Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L (2008) Rational targeting of Notch signaling in cancer. Oncogene 27:5124–5131
Rodriguez-Pascual J, Cheng E, Maroto P, Duran I (2010) Emergent toxicities associated with the use of mTOR inhibitors in patients with advanced renal carcinoma. Anticancer Drugs 21:478–486
Shepherd C, Banerjee L, Cheung CW, Mansour MR, Jenkinson S, Gale RE, Khwaja A (2012) PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia
Tolcher AW, Chugh R, Chambers G, Thorpe V, Dupont J, Hill D, Xu L, Kapoun A, Smith DC (2012) A first-in-human phase I study to evaluate the fully human monoclonal antibody OMP-59R5 (anti-Notch2/3) administered intravenously to patients with advanced solid tumors. J Clin Oncol 30:3025
Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI, Boylan JF, Xu ZX, Wang K, Koehler A, Song J, Middleton SA, Deutsch J, Demario M, Kurzrock R, Wheler JJ (2012) Phase I study of RO4929097, a Gamma secretase inhibitor of notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 30:2348–2353
Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP (2011) Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer 10:138
Wu J, Wiegand R, LoRusso P, Li J (2011) Validation and implementation of a liquid chromatography/tandem mass spectrometry assay for quantitation of the total and unbound RO4929097, a gamma-secretase inhibitor targeting Notch signaling, in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 879:1537–1543
Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464:1052–1057
Disclosures
The authors declare no conflict of interest for this manuscript.
Funding
This study (PJC-005/NCI-8500) is conducted by the Princess Margaret Hospital Phase I Consortium with support from the US National Cancer Institute U01 Cooperative Agreement Award (U01-CA132123).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 85.5 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Diaz-Padilla, I., Hirte, H., Oza, A.M. et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 31, 1182–1191 (2013). https://doi.org/10.1007/s10637-013-0001-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-0001-5