
Summary
Cytarabine (ara-C) and gemcitabine (dFdC) are commonly used anticancer drugs, which depend on the equilibrative (ENT) and concentrative-nucleoside-transporters to enter the cell. To bypass transport-related drug resistance, lipophilic derivatives elacytarabine (CP-4055), ara-C-5′elaidic-acid-ester, and CP-4126, (CO 1.01) gemcitabine-5′elaidic-acid-ester, were investigated for the entry into the cell, distribution, metabolism and retention. The leukemic CEM-cell-line and its deoxycytidine-kinase deficient variant (CEM/dCK-) were exposed for 30 and 60 min to the radiolabeled drugs; followed by culture in drug-free medium in order to determine drug retention in the cell. The cellular fractions were analyzed with thin-layer-chromatography and HPLC. Elacytarabine and CP-4126 were converted to the parent compounds both inside and outside the cell (35–45%). The ENT-inhibitor dipyridamole did not affect their uptake or retention. Inside the cell Elacytarabine and CP-4126 predominantly localized in the membrane and cytosolic fraction, leading to a long retention after removal of the medium. In contrast, in cells exposed to the parent drugs ara-C and dFdC, intracellular drug concentration increased during exposure but decreased to undetectable levels after drug removal. In the dCK- cell line, no metabolism was observed. The concentrations of ara-CTP and dFdCTP reached a peak at the end of the incubation with the drugs, and decreased after drug removal; peak levels of dFdCTP were 35 times higher than ara-CTP and was retained better. In contrast, after exposure to elacytarabine or CP-4126, ara-CTP and dFdCTP levels continued to increase not only during exposure but also during 120 min after removal of the elacytarabine and CP-4126. Levels of ara-CTP and dFdCTP were higher than after exposure to the parent drugs. In conclusion, the lipophilic derivatives elacytarabine and CP-4126 showed a nucleoside-transporter independent uptake, with long retention of the active nucleotides. These lipophilic nucleoside analogues are new chemical entities suitable for novel clinical applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The deoxynucleoside analogs ara-C and dFdC are drugs commonly used in the treatment of a variety of cancers, although ara-C is restricted for use in hematological malignancies [1, 2], while dFdC is first line therapy for non-small cell lung cancer, pancreatic cancer and bladder cancer [3–5]. Due to their hydrophilic nature these drugs are dependent on the equilibrative and concentrative nucleoside transporters (hENT and hCNT) to cross the cell membrane into the cell [6]. Both hENT and hCNT are membrane transporters required for uptake of natural nucleosides in the cell, and play an important role in the uptake of nucleoside analogs. The subsequent rate-limiting step in the activation of both drugs to its active triphosphate form is the conversion to the intermediate monophosphate by deoxycytidine kinase (dCK) [7, 8]. Both drugs can be inactivated by deoxycytidine deaminase (CDA). The secondary intermediate of dFdC, dFdC diphosphate (dFdCDP), is known to inhibit ribonucleotide reductase, which is essential in providing deoxyribonucleotides required for DNA repair [9]. DNA polymerization is stopped after incorporation of dFdC triphosphate (dFdCTP) and one more nucleotide; this masked chain termination prevents removal of dFdC by exonucleases [10]. Ara-C triphosphate (ara-CTP) incorporation into DNA also causes chain termination, while the active metabolite of ara-C can inhibit DNA polymerase by competitive inhibition [1].
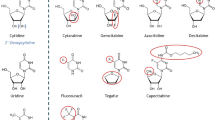
In order to enhance the uptake of the drugs into the cell by making the drugs more lipophilic, various derivatives containing a fatty acid side chain have been developed [11], which are usually coupled to the 5′ position on the sugar moiety or to the amino group of the base. The derivatives of dFdC (CP-4126, now designated as CO 1.01) and ara-C (CP-4055, elacytarabine) each contain a fatty acid at the 5′position with a chain length of eighteen carbon atoms and one trans-double bond (elaidic acid) in position 9. Elacytarabine has shown remarkable antitumor activity in various model systems including solid cancer xenografts in which the parent drug ara-C has no activity [12, 13]. CP-4126 has at least similar activity as dFdC in several xenografts models, while it also has oral activity [14]. Other studies [13, 16] have demonstrated that elacytarabine is still dependent on activation by dCK, but that the drug has a longer cellular retention and a different effect on DNA and RNA synthesis compared to ara-C. In CP-4126 the fatty acid chain also protected the dFdC from deamination [14], but CP-4126 is also dependent on dCK for activation [14, 15]. Elacytarabine was investigated in a phase I clinical study in solid tumors [17], showing a favorable and predictable safety profile which was dose and schedule dependent. Both drugs are currently in phase II studies. Other lipophilic derivatives of predominantly ara-C have shown at least similar accumulation of ara-CTP, and sometimes a prolonged retention [18]. Since some of our preliminary results indicated a delayed accumulation of the active triphosphates as well as prolonged retention, the aim of this study was to investigate uptake and metabolism of Elacytarabine and CP-4126. For this proof-of-principle study we used CEM cells and its dCK deficient variant to discriminate between uptake and metabolism.
Materials and methods
Drugs
Elacytarabine and CP-4126 (now designated as CO-1.01) were provided by Clavis Pharma (Oslo, Norway), tetrahydrouridine (THU) was from Calbiochem (Merck, Darmstadt, Germany), dipyridamole and ara-C were from Sigma-Aldrich (St. Louis, MO, USA) and dFdC was from Eli-Lilly (Indianapolis, IN, USA). The radioactively labeled drugs were obtained from Moravek (Brea, CA, USA); the drugs were labeled with tritium on the 5-C site of the base. A mix of radioactive and non-radioactive compounds was made for exposure of cells; ara-C and elacytarabine were used at a concentration of 9.7 μM and a specific activity of 1789 and 1784 mCi/mmol respectively, dFdC and CP-4126 were used at a final concentration of 8.9 μM and a specific activity of 586 and 391 mCi/mmol, respectively.
Cell lines
For the experiments the CCRF-CEM human leukemia cell line and its dCK negative variant (CEM/dCK-) were used. CEM/dCK- cells are completely resistant to both ara-C and dFdC and their prodrugs, due to the absence of dCK. IC50 values are given in Table 1. The latter cell line was required to get a clean drug uptake and sequestration pattern, not affected by activation of the parent drug. The cell lines were cultured in RPMI medium (BioWhittaker, Verviers, Belgium) supplemented with 10% fetal calf serum (PAA laboratories, Pasching, Austria) and HEPES buffer (BioWhittaker). Of the nucleoside influx transporters, the CEM cell line only expresses hENT and not hCNT [19]. Dipyridamole was used to inhibit the influx of the drugs by hENT. In order to get a clean picture we also added THU to inhibit deamination of ara-C and dFdC; hence the only metabolic pathway available for the drugs is activation by dCK.
Drug uptake and metabolism
The method was based on a procedure described earlier [20]. Shortly, cells were harvested and resuspended in fresh medium at 5 × 106 cells/ml. Of this cell suspension 100 μl was used for each experiment. To inhibit deamination by CDA, THU was added at a final concentration of 100 μM and was present during incubation and when incubated in drug free medium, which is sufficient to keep deamination inhibited [21]; the drugs were added to reach a final concentration of 9.7 and 8.9 μM for ara-C/elacytarabine and dFdC/CP-4126, respectively. The cells were incubated for 0, 30 and 60 min at 37°C. To measure intracellular drug retention the drug containing medium was replaced after 60 min and the cells were incubated for 60 min in drug free medium. Thereafter the cells were spun down (3000 g, 2 min, 4°C) and the medium was stored as extracellular fraction at −20°C. The cells were washed with cold PBS (12000 g, 1 min, 4°C). The cell pellet was resuspended in 45 μl cold PBS and nucleosides and nucleotides were extracted by addition of 5 μl perchloric acid (5 M) and chilled on ice for 20 min. After spinning down (12000 g, 3 min, 4°C) the perchloric acid pellet containing the precipitated nucleic acids was resuspended in 200 μl NaOH (1 M). The supernatant containing the cytosolic fraction was neutralized with 10 μl KH2PO4 (5 M) and stored as intracellular fraction at −20°C. Using 3H-elacytarabine and 3H-CP-4126 we did not find evidence that the prodrugs were hydrolyzed to ara-C or dFdC under acidic conditions used for precipitation of proteins and nucleic acids.
Of the extracellular and intracellular cytosolic samples 5 μl was spotted on a plastic backed silica TLC plate (Merck KgaA, Darmstadt, Germany) in order to separate ara-C from elacytarabine and formed nucleotides and to separate dFdC from CP-4126 and formed nucleotides. The chromatography was performed with 3:2 chloroform/methanol as a mobile phase. After separation the spots were visualized with UV light and cut into separate scintillation vials, and radioactivity was eluted by overnight incubation in methanol. The samples were measured together with the perchloric acid pellet samples in an LSC counter. All experiments were performed in duplicate and repeated at least 3 times.
Intracellular localization
Intracellular localization of the lipophilic analogs was investigated using a ProteoExtract™ Subcellular Proteome Extraction Kit (Calbiochem). Cells were incubated with 9.7 and 8.9 μM of the radioactive drugs as described above and deamination was inhibited by 100 μM THU. The samples were incubated for 60 min at 37°C. After incubation the cells were washed and with the different reagents provided with the kit; the samples were separated into subcellular fractions: a cytosolic, a membrane, a nuclear and a cytoskeletal fraction. All experiments were performed in duplicate and repeated at least 3 times. Differences were evaluated using the Student’s t-test.
Triphosphate accumulation
Cells were treated with 1 and 10 μM of dFdC and ara-C, respectively, while for the lipophilic analogs higher concentrations of 10 and 100 μM was used, since initial experiments demonstrated that this was required to achieve similar triphosphate accumulation. The cells were incubated for 60 min and retention of the triphosphates was investigated after 60 and 120 min incubation in drug-free medium. Dipyridamole was added to inhibit drug influx and efflux by the hENT transporter. After incubation the cells were spun down (12000 g, 1 min, 4°C) and the cell pellet was resuspended in ice-cold PBS and incubated for 20 min at 4°C with 40% trichloroacetic acid to extract the nucleotides. After centrifugation (10,000 g, 10 min, 4°C) the supernatant was neutralized by addition of a 2-fold excess of trioctylamine/1,1,2-trichlorotrifluorethane (1:4) and spun down (10,000 g, 1 min) and the aqueous phase containing the extracted nucleotides was stored at −20°C until analysis by HPLC on a Whatman Partisphere SAX column (GE healthcare, Chalfont St. Giles, UK) using either isocratic (to separate ara-CTP from normal nucleotides and quantifiy ara-CTP) or gradient (to separate dFdCTP from other nucleotides and quantify dFdCTP) elution as described earlier [22, 23].
Results
Incubation of the CEM and CEM/dCK- cell lines with elacytarabine led to release of free ara-C in the extracellular medium, which increased over time in both cell lines (Fig. 1). After exposure of the CEM cell line to ara-C, the levels of ara-C decreased gradually while the levels remained the same in the CEM/dCK- cell line. Incubation with elacytarabine followed by culturing in drug free medium led to higher extracellular levels of ara-C than after incubation with ara-C.

Extracellular concentration of ara-C and dFdC after incubation for 30 or 60 min (marked as 30 and 60) of the CEM or CEM/dCK- cell lines with radioactive drugs. The concentrations of the prodrug and parent drug were measured after 60 min exposure to the drugs and washing with drug-free medium and culture for 60 min in drug free medium (marked at 60 + 60). a Ara-C after incubation with 9.7 μM [3H]-ara-C or [3H]-elacytarabine (CP-4055), b dFdC after incubation with 8.9 μM [3H]-dFdC or [3H]-CP-4126. Values represent means ± SEM of at least three separate experiments. Ara-C levels in medium of CEM-dCK- cells were significantly higher than in medium of CEM cells (P < 0.01)
In contrast to elacytarabine, extracellular CP-4126 was converted very rapidly to dFdC by both cell lines; after 30 min incubation almost all the extracellular CP-4126 was converted to dFdC (Fig. 1). After incubation of the CEM cell line with dFdC the levels decreased over time, although this was not significant, while in the CEM/dCK- cell line the levels remained the same. The dFdC formed from CP-4126 showed the same trend as the normal dFdC levels, some dFdC was left in the medium after removal of the drugs. In contrast to data with elacytarabine, accumulation of the dFdC after removal of the drug was limited.
After incubation with elacytarabine, intracellular ara-C accumulated faster and to higher levels compared to incubation with ara-C (Fig. 2). After removal of elacytarabine, ara-C continued to be released inside the cells. After 60 min incubation with elacytarabine, the extent of ara-C accumulation was twice as high as after incubation with ara-C. The absence of dCK did not affect the initial accumulation of ara-C after incubation with elacytarabine, but after 60 min less ara-C was present in the CEM/dCK- cells than in the wild type CEM cell line. This difference is most likely caused by the trapping of ara-C in nucleotides in the CEM cells, but not in the CEM/dCK- cells (see below). Intracellular dFdC formed from CP-4126 showed a different accumulation profile as for dFdC itself (Fig. 2). Levels of dFdC formed from CP-4126 were 10-times higher compared to that after incubation with dFdC alone and peaked after 30 min incubation, after which the levels decreased and were comparable for dFdC from CP-4126 and from dFdC itself. Similarly, levels of dFdC from CP-4126 were higher (6-times) than that from dFdC in the CEM/dCK-. However, dFdC levels were lower than in the CEM cell line, possibly because of lack of metabolism in the CEM/dCK- cells. No differences in accumulation after removal of the drug between dFdC and dFdC formed from CP-4126 were observed.

Intracellular accumulation of ara-C and dFdC after incubation for 30 or 60 min of the CEM or CEM/dCK- cell line with radioactive drugs (marked as 30 and 60). Retention of the prodrug and the parent drug was measured after exposure for 60 min and washing with drug-free medium and culture for 60 min in drug free medium (marked as 60 + 60). a Ara-C after incubation with 9.7 μM [3H]-ara-C or [3H]-elacytarabine (CP-4055), b dFdC after incubation with 8.9 μM [3H]-dFdC or [3H]-CP-4126. Values represent means ± SEM of at least three separate experiments. Accumulation of ara-C from CP-4055 after 60 min and 60 min retention period was significantly higher in CEM-cells than in CEM-dCK- cells, similarly accumulation of dFdC from CP-4126 after 30 min was significantly higher in CEM compared to CEM-dCK- cells (P < 0.05)
Exposure to radiolabelled ara-C led to some ara-C accumulation in the nuclear pellet of CEM cells representing ara-C incorporation into DNA, but in the CEM/dCK- cell line the accumulation of ara-C did not exceed the background in line with the absence of dCK (Fig. 3). However, after incubation with elacytarabine at least 100 times higher accumulation of radioactivity in the perchloric acid pellet was observed than with ara-C. In contrast to incubation with ara-C, the accumulation of radioactivity after incubation with elacytarabine was similarly high in CEM and CEM/dCK- cells and did not change over time. Radioactivity in the pellet was retained at a similar level in the pellet of CEM/dCK- and CEM cells, indicating that this radioactivity most likely does not represent ara-C incorporation into DNA. After incubation with 3H-dFdC accumulation of radioactivity in the nuclear pellet, representing 3-H-dFdC incorporation into DNA, showed the same trend as for 3H-ara-C (Fig. 3). However, radioactive CP-4126 showed a high accumulation in the perchloric acid pellet of the CEM and CEM/dCK- cell lines, which decreased over time. This decrease was more rapid in CEM cells compared to CEM/dCK- cells, and more rapid then for elacytarabine.

Nuclear accumulation and retention of radiolabeled ara-C (a), elacytarabine (CP-4055) (b), dFdC (c) and CP-4126 (d). The nuclear pellets of CEM and CEM/dCK- cells were analyzed for radiolabeled metabolites after incubation for 30 or 60 min (marked as 30 and 60) with 9.7 μM [3H]-ara-C (a) or [3H]-elacytarabine (CP-4055) (b), 8.9 μM [3H]-dFdC (c) or [3H]-CP-4126 (d). Retention was studied after 60 min exposure followed by washing the cells and resuspension in drug-free medium for 60 min (marked as 60 + 60). Values represent means ± SEM of at least three separate experiments. Accumulation of radiolabeled CP-4126 was significantly lower than that of CP-4055 (P < 0.01)
In order to verify that the difference in extracellular concentration and intracellular accumulation of ara-C after incubation with [3H]-elacytarabine or [3H]-ara-C was due to hENT mediated transport for ara-C and not for elacytarabine, we added dipyridamole to the cell cultures. The extracellular concentration of ara-C did not change in time and was almost undetectable after washing the cells with drug-free medium. Consequently no intracellular ara-C was found after incubation with dipyridamole, also precluding any passive diffusion of ara-C. However, dipyridamole did not affect the accumulation of ara-C released from elacytarabine, indicating that elacytarabine enters the cell independently of the hENT transporter (Fig. 4), and that ara-C is released from elacytarabine inside the cell. Similar data were found for dFdC and CP-4126 (data not shown), in which dipyridamole completely inhibited dFdC uptake, excluding passive diffusion of dFdC, but did not affect uptake of CP-4126 and subsequently release of dFdC.

Effect of dipyridamole on (a) Extracellular concentration and (b) intracellular accumulation of ara-C and elacytarabine (CP-4055) in CEM cells (marked as 30 and 60). Cells were incubated for 30 and 60 min with 9.7 μM [3H]-ara-C or [3H]-elacytarabine (CP-4055), after which elacytarabine and ara-C were measured. Concentrations of the prodrug and parent drug were measured after 60 min exposure and washing the cells with drug free medium and incubation for 60 min in drug free medium (marked as 60 + 60). Values represent means ± SEM of at least three separate experiments
Since the perchloric acid procedure not only precipitated ara-C and dFdC incorporated into DNA, but also additional elacytarabine and CP-4126, this precluded measurement of ara-C and dFdC incorporation into DNA after exposure to elacytarabine or CP-4126. In order to get more insight in the formation of active metabolites formed from the prodrugs we therefore determined the accumulation of triphosphates (ara-CTP and dFdCTP). After 60 min incubation with ara-C or dFdC, more triphosphates were formed than after incubation with elacytarabine or CP-4126, respectively (Fig. 5). The concentrations of ara-CTP and dFdCTP formed from ara-C or dFdC decreased after washing away the drugs but the concentration of ara-CTP from elacytarabine continued to increase while dFdCTP from CP-4126 was retained at a similar level. In order to completely block carrier-mediated transport we added the ENT inhibitor dipyridamole to the assay. CEM cells do not express CNTs which may also transport dFdC. Dipyridamole completely inhibited entry of the parent compounds, abolishing accumulation of ara-CTP and dFdCTP. However, dipyridamole even increased the concentration of triphosphates from the prodrugs even after washing away the drugs. Earlier we demonstrated that CEM/CK- cells do not phosphorylate deoxycytidine analogs because of the dCK deficiency [24]. Neither ara-C or Elacytarabine were phosphorylated to detectable levels in the CEM/dCK- cells [24], since Ara-C can only be phosphorylated by dCK. Although alternative kinases such as thymidine kinase 2 can phosphorylate dFdC, but we could not detect any dFdCTP in the CEM/dCK- cells. Even using a more sensitive LC-MS-MS assay for all dFdC-phosphates [25], did not give evidence for formation of any dFdC nucleotide in the CEM/dCK- cells, while dFdC-phosphates were easily detectable in wild-type cells.

Ara-CTP (a) and dFdCTP (b) accumulation in the CEM cell line after 60 min incubation with 10 μM ara-C, 100 μM elacytarabine (CP-4055), 1 μM dFdC or 10 μM CP-4126 with and without inhibition of hENT by dipyridamole. Intracellular concentrations of the triphosphates were measured after 60 min exposure and washing the cells with drug-free medium and subsequent culture for 60 and 120 min in drug-free medium (marked as 60 + 60 and 60 + 120). Values represent means ± SEM of at least three separate experiments. Accumulation (60 min) of ara-CTP and dFdCTP was not different when comparing ara-C incubation with elacytarabine or dFdC with CP-4126. However, the retention (60 + 120) of ara-CTP and of dFdCTP was significantly higher in cells incubated with elacytarabine (p < 0.01) or CP-4126 (p < 0.05), respectively, compared to incubation with ara-C or dFdC, as well as the effect of dipyridamole on dFdCTP retention (P < 0.01) of ara-CTP retention (P < 0.05)
Because of the unexpected high accumulation of radioactivity in the perchloric acid pellet of both CEM and CEM/dCK- cells, we assumed an accumulation of the lipophilic analogs in subcellular fractions, either protein-bound or in lipid vesicles, which were precipitated by perchloric acid. Since this might also explain the longer retention of the lipophilic analog in the cell, we investigated the subcellular distribution of the drugs (Fig. 6). The more elaborate procedure enabled to analyze accumulation in different cellular compartments. In the parent CEM cell line both the lipophilic analogs and the nucleoside analogs accumulated predominantly in the cytosolic fraction, while a considerable amount of elacytarabine was found in the membrane fraction. However, in the CEM/dCK- cell line the relative accumulation of the lipophilic analogs in the membrane fraction was much higher, as well as in the nucleus, possibly because of a lack of metabolism in these cells. This fits very well with the high accumulation of the labeled Elacytarabine and CP-4126 in the nuclear fraction as found with perchloric acid precipitation

Subcellular localization of radioactivity from 9.7 μM Elacytarabine (CP-4055) (a,b) and ara-C (a,b), or 8.9 μM CP-4126 (c,d) and dFdC (c,d) in the CEM (a,c) and CEM/dCK- (b,d) cell lines. Relative amounts after 60 min incubation with the radioactive drugs; total activity was calculated by setting the sum of radioactivity in all fractions at 100%, and subsequently calculating the percentage in each fraction. Accumulation of elacytarabine label and CP-4126 label was significantly higher in the membrane fraction compared to accumulation of ara-C (p < 0.05) or dFdC (p < 0.02) label, respectively
Discussion
The lipophilic analogs CP-4126 (gemcitabine–5′-elaidic acid ester) and elacytarabine (cytarabine–5′-elaidic acid ester) showed differential effects in metabolism and accumulation not only compared to their respective parent compounds but also compared to each other. The prodrugs seem to be trapped in an intracellular compartment, subsequently releasing the prodrug itself or the parent compound. Both analogs were converted by a non-identified esterase to their parent compounds outside but predominantly inside the cell.
In the medium CP-4126 disappeared very rapidly and was converted to dFdC but accumulation in cells of both dFdC and partly dFdCTP was higher than for dFdC itself. The difference in conversion rate between elacytarabine and CP-4126 is probably caused by the fluorine atoms in the sugar of the dFdC part of the analog (Table 2). The longer bond length for CP-4126 between the C and O atoms indicates a weaker binding, which might explain the more rapid hydrolysis of CP-4126 to dFdC relative to elacytarabine hydrolysis to ara-C [26]. Although this does not seem to affect the in vivo efficiency of the drug it does cause the compound to be converted rapidly in the experimental setup of our experiments.
The most pronounced difference between prodrug and parent compound was observed for elacytarabine and ara-C. Ara-C itself was not retained for a long period, similar to other studies with ara-C in leukemic cell lines and patient samples [27, 28]. However, when cells were exposed to elacytarabine, ara-C continued to be released, even after incubation in drug-free medium. This was also reflected in ara-CTP accumulation, which after incubation with elacytarabine continued to increase when elacytarabine was washed away, in contrast to ara-CTP from ara-C, which decreased rapidly during incubation in drug-free medium [22]. Elacytarabine was shown to enter the cell independently of the hENT transporter, thereby circumventing a possible resistance mechanism to ara-C, confirming previous results [12, 29]. Inhibition of the hENT transporter caused an increased accumulation of both ara-CTP and dFdCTP from the lipophilic analogs. The reason for this effect might be the specific inhibition of hENT, which also catalyzes efflux of ara-C and dFdC, which accumulated in the cell after being released from the prodrugs. Since CEM cells do not express CNTs, under these conditions only diffusion may play a role in influx and efflux of nucleosides, but nucleotides are too polar to diffuse out of the cells. Although BCRP and MRP-4 may also be inhibited by dipyridamole, the inhibition of hENT seemed to be the most prominent effect, since no uptake of ara-C or dFdC was observed. An inhibitory effect of dipyridamole on efflux of the mononucleotides of elacytarabine and CP-4126 may theoretically be part of the explanation as well.
Also dFdC released from CP-4126 rapidly reached higher concentrations of dFdC than when cells were incubated with dFdC. Subsequently this increase in dFdC from CP-4126 led to a high accumulation of dFdCTP. In contrast to ara-CTP, dFdCTP elimination after dFdC exposure is biphasic and much slower [9, 23]. The retention of dFdCTP from CP-4126 even seemed to be longer. The higher sensitivity of the CEM wild type cells to dFdC and CP-4126 compared to ara-C and Elacytarabine is reflected in the higher accumulation of dFdCTP compared to ara-CTP. Unfortunately, the prodrugs were not able to bypass resistance to ara-C and dFdC in CEM/dCK- cells. Both Elacytarabine and CP-4126 do not contain a phosphate between the sugar and elaidic acid (Table 2); therefore they can only be broken down to the nucleoside analogs ara-C and dFdC and elaidic acid. Despite the higher accumulation of dFdC in CEM/dCK- cells, these cells can not phosphorylate ara-C or dFdC, which is due to the deficiency of dCK. Although dFdC is also a substrate for other kinases such as thymidine kinase 2 [7], their activity is apparently too low in these cells to catalyze the formation of detectable levels of dFdC nucleotides.
The differences between lipophilic analogs and parent compounds can at least partly be explained by the intracellular localization of the compounds. The lipophilic fatty acid chain attached to the drug caused the drug to be retained in a membrane fraction inside the cell. This accumulation, possibly in a membrane vesicle, caused a slower and prolonged release of the nucleoside analog from the lipophilic analogs. This ultimately caused an effect that is different from the nucleoside analogs from which the lipophilic analogs are derived. This differential effect of the lipophilic analogs compared to the parent compounds shows that these prodrugs have a different mechanistic effect in cells [11]. The intracellular sequestration of the lipophilic prodrugs in the cell may have profound implications for the clinical application of these drugs. This prolonged intracellular accumulation of the prodrugs and their metabolites will lead to a different drug behavior compared to ara-C and dFdC themselves. The antitumor activity of both ara-C and dFdC is dependent on the duration of retention of the active metabolite. Therefore the longer retention of the prodrug and of the triphosphates represents a favorable property compared to the parent drug and offers promise to improve treatment of malignancies currently being treated with ara-C or dFdC, and to treat other malignancies with these drugs. Since accumulation of ara-CTP has been shown to be a valuable pharmacodynamic biomarker for the efficacy of ara-C in leukemia, these levels should be monitored in clinical studies with Elacytarabine, while those of dFdCTP should be measured in studies with CP-4126.
Both the independence on influx transporters and the continuing accumulation and retention after removal of the drugs, create the possibility for novel combinations and treatment in tumors where the parent compounds show less or no effect.
Abbreviations
- ara-C:
-
Cytarabine
- ara-CTP:
-
Ara-C triphosphate
- CP-4055:
-
Elacytarabine, cytarabine–5′-elaidic acid ester
- CP-4126, CO-101:
-
Gemcitabine–5′-elaidic acid ester
- dFdC:
-
Gemcitabine
- dFdCTP:
-
dFdC triphosphate
- dCK:
-
Deoxycytidine kinase
- CDA:
-
Cytidine deaminase
- CEM/dCK-:
-
dCK negative variant of CEM
- hCNT:
-
Human concentrative nucleoside transporter
- hENT:
-
Human equilibrative nucleoside transporter
- PBS:
-
Phosphate buffered saline
- THU:
-
Tetrahydrouridine
References
Plunkett W, Gandhi V (1993) Cellular pharmacodynamics of anticancer drugs. Semin Oncol 20:50–63
Rustum YM, Raymakers RA (1992) 1-Beta-arabinofuranosylcytosine in therapy of leukemia: preclinical and clinical overview. Pharmacol Ther 56:307–321
Ramalingam S, Belani C (2008) Systemic chemotherapy for advanced non-small cell lung cancer: recent advances and future directions. Oncologist 13(Suppl 1):5–13
Heinemann V (2002) Gemcitabine in the treatment of advanced pancreatic cancer: a comparative analysis of randomized trials. Semin Oncol 29:9–16
Hussain SA, James ND (2003) The systemic treatment of advanced and metastatic bladder cancer. Lancet Oncol 4:489–497
Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE (1998) Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58:4349–4357
Bergman AM, Pinedo HM, Peters GJ (2002) Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug Resist Updat 5:19–33
Hubeek I, Kaspers GJ, Ossenkoppele GJ, Peters GJ (2006) Cytosine arabinoside. Metabolism, mechanisms of resistance and clinical pharmacology. In: Peters GJ (ed) Cancer drug discovery and development: deoxynucleoside analogs in cancer therapy. Humana Press inc, Totowa, pp 119–152
Plunkett W, Huang P, Searcy CE, Gandhi V (1996) Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol 23:3–15
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W (1991) Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 51:6110–6117
Adema AD, Bijnsdorp IV, Sandvold ML, Verheul HM, Peters GJ (2009) Innovations and opportunities to improve conventional (deoxy) nucleoside and fluoropyrimidine analogs in cancer. Curr Med Chem 16:4632–4643
Breistol K, Balzarini J, Sandvold ML, Myhren F, Martinsen M, De Clercq E, Fodstad O (1999) Antitumor activity of P-4055 (elaidic acid-cytarabine) compared to cytarabine in metastatic and s.c. human tumor xenograft models. Cancer Res 59:2944–2949
Bergman AM, Kuiper CM, Voorn DA, Comijn EM, Myhren F, Sandvold ML, Hendriks HR, Peters GJ (2004) Antiproliferative activity and mechanism of action of fatty acid derivatives of arabinofuranosylcytosine in leukemia and solid tumor cell lines. Biochem Pharmacol 67:503–511
Bergman AM, Adema AD, Balzarini J, Bruheim S, Fichtner I, Noordhuis P, Fodstadt Ø, Myhren F, Sandvold ML, Hendriks H, Peters GJ (2011) Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs 29:456–466
Bergman AM, Kuiper CM, Noordhuis P, Smid K, Voorn DA, Comijn EM, Myhren F, Sandvold ML, Hendriks HR, Fodstad O, Breistol K, Peters GJ (2004) Antiproliferative activity and mechanism of action of fatty acid derivatives of gemcitabine in leukemia and solid tumor cell lines and in human xenografts. Nucleosides Nucleotides Nucleic Acids 23:1329–1333
Adema AD, Laan AC, Myhren F, Fichtner I, Verheul HM, Sandvold ML, Peters GJ (2010) Cell cycle effects of fatty acid derivatives of cytarabine, CP-4055, and of gemcitabine, CP-4126, as basis for the interaction with oxaliplatin and docetaxel. Int J Oncol 36:285–294
Dueland S, Aamdal S, Lind MJ, Thomas H, Sandvold ML, Gaullier JM, Rasch W (2008) Intravenous administration of CP-4055 (ELACYTtrade mark) in patients with solid tumours. A phase I study. Acta Oncol 48:137–145
Horber DH, Schott H, Schwendener RA (1995) Cellular pharmacology of N4-hexadecyl-1-beta-D-arabinofuranosylcytosine in the human leukemic cell lines K-562 and U-937. Cancer Chemother Pharmacol 36:483–492
Belt JA, Marina NM, Phelps DA, Crawford CR (1993) Nucleoside transport in normal and neoplastic cells. Adv Enzyme Regul 33:235–252
Peters GJ, Laurensse E, Lankelma J, Leyva A, Pinedo HM (1984) Separation of several 5-fluorouracil metabolites in various melanoma cell lines. Evidence for the synthesis of 5-fluorouracil-nucleotide sugars. Eur J Cancer Clin Oncol 20:1425–1431
Yusa K, Oh-hara T, Tsuruo T (1995) Induction of resistance to 1-beta-D-arabinofuranosylcytosine in human H9 cell line by simian immunodeficiency virus. Biochem Biophys Res Commun 206:486–491
Noordhuis P, Kazemier KM, Kaspers GJ, Peters GJ (1996) Modulation of metabolism and cytotoxicity of cytosine arabinoside with N-(phosphon)-acetyl-L-aspartate in human leukemic blast cells and cell lines. Leuk Res 20:127–134
Ruiz van Haperen V, Veerman G, Boven E, Noordhuis P, Vermorken JB, Peters GJ (1994) Schedule dependence of sensitivity to 2′,2′-difluorodeoxycytidine (Gemcitabine) in relation to accumulation and retention of its triphosphate in solid tumour cell lines and solid tumours. Biochem Pharmacol 48:1327–1339
Adema AD, Loosekoot N, Smid K, Kathmann I, Myhren F, Sandvold ML, Peters GJ (2010) Induction of resistance to the lipophilic cytarabine prodrug Elacytarabine (CP-4055) in CEM leukemic cells. Nucleosides, Nucleotides and Nucleic Acids 29:394–399
Honeywell R, Laan AC, Van Groeningen CJ, Strocchi E, Ruiter R, Giaccone G, Peters GJ (2007) The determination of gemcitabine and 2′-deoxycytidine in human plasma and tissue by APCI tandem mass spectrometry. J Chromatog B 847:142–152
Hausheer FH, Jones ND, Seetharamulu P, Singh UC, Deeter JB, Hertel LW, Kroin JS (1996) Ab initio quantum mechanical and X-ray crystallographic studies of gemcitabine and 2′-deoxy cytosine. Comput Chem 20(4):459–467
Plunkett W, Liliemark JO, Adams TM, Nowak B, Estey E, Kantarjian H, Keating MJ (1987) Saturation of 1-beta-D-arabinofuranosylcytosine 5′-triphosphate accumulation in leukemia cells during high-dose 1-beta-D-arabinofuranosylcytosine therapy. Cancer Res 47:3005–3011
Preisler HD, Rustum Y, Priore RL (1985) Relationship between leukemic cell retention of cytosine arabinoside triphosphate and the duration of remission in patients with acute non-lymphocytic leukemia. Eur J Cancer Clin Oncol 21:23–30
Galmarini CM, Myhren F, Sandvold ML (2008) CP-4055 and CP-4126 are active in ara-C and gemcitabine-resistant lymphoma cell lines. Br J Haematol 144:273–275
Acknowledgements
This study was supported by an educational grant from Clavis Pharma, Oslo, Norway.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Financial support and conflict of interest
Financial support was received from Clavis Pharma to pay one of the researchers (ADD). MLS and FM are employees of Clavis Pharma, NK, KS, RJH, HMV and GJP from the VU University Medical Center.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Adema, A.D., Smid, K., Losekoot, N. et al. Metabolism and accumulation of the lipophilic deoxynucleoside analogs elacytarabine and CP-4126. Invest New Drugs 30, 1908–1916 (2012). https://doi.org/10.1007/s10637-011-9756-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-011-9756-8