
Abstract
Population bottlenecks may result in the loss of genetic diversity, with potentially negative consequences for species of interest in conservation biology, including rare species, invasive species and biological control agents. We examined mtDNA sequence data and four variable microsatellite loci (SSRs) in the melaleuca psyllid Boreioglycaspis melaleucae, which was introduced from Australia to Florida as a biological control agent of the invasive plant Melaleuca quinquenervia. We sampled psyllids in the native and introduced ranges as well as individuals stored from the original founding population. There was a clear loss of mtDNA haplotype diversity, as well as a loss of rare microsatellite alleles, in the introduced range. However, there was little genetic differentiation between the home and introduced ranges, and no evidence for a genetic bottleneck based on an analysis of heterozygosity with the microsatellite markers. Overall, the data showed that the demographic bottleneck had a limited effect on the genetics of populations in the new range.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
When species colonize a new location, a relatively small number of individuals may found the new populations (Elton 1958; Debach and Rosen 1991). This bottleneck may result in a loss of genetic diversity (Nei et al. 1975; Templeton 1980; Barton and Charlesworth 1984; Hartl and Clark 2007), which can limit the evolutionary potential of the populations (Avise and Hamrick 1996; Frankham and Ballou 2003). This evolutionary potential can be particularly critical to success in a novel environment (Cox 2004).
Genetic bottlenecks can occur in species intentionally released as biological control agents against invaders, since they are often collected in limited quantities from a small number of sites (Debach and Rosen 1991). Genetic diversity may be further reduced due to mortality in transit and inbreeding in quarantine. While many biological control agents have established, others have not been as successful (Debach and Rosen 1991; Pemberton 2000; Louda et al. 2003; Culliney 2005). Loss of genetic diversity could be one factor limiting successful establishment of biological control agents.
Most studies of genetic bottlenecks attempt to infer demographic factors such as the minimum population size and duration of the reduction based on genetic data (Chen et al. 2006; Barker et al. 2009). Some approaches assume that following a demographic bottleneck, allelic diversity is reduced faster than heterozygosity, so that a bottleneck can be inferred if observed heterozygosity is larger than expected at mutation-drift equilibrium given a particular mutation model (Cornuet and Luikart 1996). Other approaches assume that the largest effect of a bottleneck is a loss of rare alleles (Slatkin 1985). However, it is difficult to test assumptions and relative merits of these approaches unless the actual demographic information is known.
Work on intentionally introduced species such as biological control agents can often take advantage of a large amount of information on the timing, duration and intensity of a demographic bottleneck because of records of the introduction (Debach and Rosen 1991; Hufbauer et al. 2004; Goolsby et al. 2006). We took advantage of a unique opportunity to investigate the population genetics of an introduced species with a very well characterized introduction history and with genetic material available from the original founding population. The melaleuca psyllid Boreioglycaspis melaleucae (Moore) (Hemiptera: Psyllidae) is a sap-feeding insect that was introduced into Florida in 2002 to aid in the control of the tree Melaleuca quinquenervia (Cav.) Blake (melaleuca), which is originally from Australia and is one of the most invasive species in the Florida Everglades (Purcell et al. 1997; Serbesoff-King 2003; Wineriter et al. 2003). Not only are the precise collection locations in Australia and introduction locations in Florida known, there were preserved insects from the population that had been transferred through a quarantine facility, the sole point of entry, prior to release.
We sampled adult psyllids from natural populations throughout the native range of Australia and the introduced range in Florida, as well as from individuals stored in quarantine that were originally used as the founding populations (Table 1). We extracted nuclear and mitochondrial DNA (mtDNA) from individual adult psyllids and sequenced the nuclear DNA for highly variable microsatellite (SSR) markers and mtDNA for sequence variation in a known variable region. We analyzed spatial genetic variation and population genetic structure within and across populations and continents to examine the genetic consequences of the introduction of this biological control agent.
Materials and methods
Study species
We investigated population genetics of the insect B. melaleucae, the melaleuca psyllid. Host range testing showed that B. melaleucae feed only on M. quinquenervia and three other closely related species in the M. leucadendra complex (Purcell et al. 1997; Wineriter et al. 2003). The psyllids feed on the sap of leaves of melaleuca seedlings and mature trees, and excrete a distinctive white flocculence. This feeding reduces plant growth and survival (Franks et al. 2006; Morath et al. 2006; Center et al. 2006; Rayamajhi et al. 2008; Tipping et al. 2008). The psyllids pass through distinctive life stages, developing from eggs, generally laid on the margins of leaves, to first through fifth instar nymphs. In the final stage of development, they become alate adults. The time for one generation is about 1 month under ideal conditions. The adults mate sexually, and there is no known parthenogenesis in this species.
For the introduction of this species, colonies of B. melaleucae were collected from six sites that were each less than 100 km from Brisbane, Australia between May 1997 and December 2001 (Center et al. 2006). The number of individuals collected per site ranged from ten to several hundred and included all life stages. From these collections, six separate shipments of the psyllid were made to the quarantine facility in Gainesville, Florida, where the insects were maintained for host range testing (Wineriter et al. 2003; Center et al. 2006). From this quarantine population, a total of 45,965 individuals were released to south Florida during the period of February 12 through April 18, 2002. Adult psyllids were originally released in 8 Melaleuca stands that were widely distributed across the range of the invasive tree in south Florida (Center et al. 2006). From these initial release sites, B. melaleucae dispersed at a rate of 4.7 km/year (Center et al. 2006) but additional redistribution efforts resulted in the movement of over two million B. melaleucae individuals from these original release sites to new Melaleuca stands that occurred outside the natural dispersal abilities of the insect.
Sampling design
In 2004 (2 years post-introduction), we collected B. melaleucae individuals from the native range of Australia and the introduced range in Florida, as well as from the original release population that had been stored in quarantine. The collection sites (populations) are shown in Table 1. There were 11 collection sites in Australia, 11 in Florida and six colonies from quarantine that represented unique collections from the source locations. The sampling locations were throughout the current range of the psyllid in both Australia and Florida. From each population, two individuals were used for analysis of mtDNA and up to 20 were used for analysis of microsatellites. The psyllids collected were all mature adults, and both males and females were included in the sampling.
Genetics
For microsatellite development and population genetic analysis, DNA was extracted from whole wild-collected adult psyllids using the ZR genomic DNA kit (Zymo Research, Orange, California) following the solid tissue protocol. In the mtDNA sequence analysis, two individuals from each of 28 populations were amplified using PCR. An approximately 650 bp fragment spanning the COI-tRNA-COII region was generated using the primers UEA9 (Lunt et al. 1996) and C2-N-3389 (Simon et al. 1994). The PCR conditions were denaturation at 94°C for 5 min, 41 cycles of denaturation at 94°C for 30 s, annealing at 48°C for 30 s, and extension at 70°C for 30 s, with a final extension of 70°C for 10 min. Some amplifications required reducing the annealing temperature to 45°C and four additional cycles for a total of 45. Amplified products were purified prior to sequencing by incubation with 3μ Exonuclease and 2μ Shrimp Alkaline Phosphatase for 30 min at 37°C, followed by 15 min at 80°C for enzyme deactivation. Products were sequenced from both ends using the same primers and BigDye Terminator Cycle Sequencing Kit v3.1, and were run on an ABI3730xl automated sequencer (Applied Biosystems, Foster City, CA). Sequences were edited and aligned using Sequencher (GeneCodes, Ann Arbor, MI) and trimmed to the same length.
Microsatellite markers were developed by the genetics facility at the Savannah River Ecology Laboratory following techniques described in Glenn and Schable (2005). Twenty-two microsatellite loci were identified (Suppl. 1). Of these, only four were shown to have clean amplification and variability in sample populations and were used in the analyses (Table 2).
We amplified the microsatellite loci in 10 μl PCR reactions, using either 0.5 or 1.0 μl of template DNA, following a program of 1–2-min cycle at 94°C; 30 cycles of 92°C for 30 s, 58°C for 1 min, and 72°C for 45 s; one cycle at 72°C for 3 min. The amplicons were separated and visualized on an ABI 3100 Genetic Analyzer (Applied Biosystems) using ABI ROX500 as a reference, and genotype scoring was performed using STR and (Hughes 1998). Because microsatellite genotyping was performed on DNA extracts from whole insects, it is possible that DNA from stored male sperm may have also been present in extractions from adult females and that samples included not only mtDNA from the target individual but also from mtDNA in the stored sperm. However, we did not observe low-intensity allele bands as sperm genotyping would produce. Further, we did not find any samples that possessed multiple different mtDNA haplotypes, which would be expected if sperm were sampled and the sperm was derived from fertilizations from different males.
Statistical analyses
We analyzed population genetic parameters and the genetic consequences of the demographic bottleneck in several ways. With the microsatellite data, allele frequencies and genetic diversity statistics were calculated with Arlequin 3.1 (Excoffier et al. 2005). These parameters were compared among the Australia, Florida and quarantine populations using analysis of variance (ANOVA) in SPSS (version 12), with pairwise post-hoc comparisons made using the Least Significant Difference (LSD) test. The Garza and Williamson (G–W) statistic (Garza and Williamson 2001; Excoffier et al. 2005), also calculated with Arlequin 3.1, is the ratio between allele size and range. This statistic is expected to reflect a recent bottleneck because the number of alleles should be more reduced than allelic range, such that G–W should be near zero in recently bottlenecked populations and near one in stable populations (Garza and Williamson 2001). Population structure statistics were computed using Fstat 2.9.3.2 (Goudet 1995). This program estimates F-statistics following Weir and Cockerham (1984). We performed a hierarchical AMOVA, which partitions genetic variance into among source continents, among populations, among and within individual components. We also looked for evidence of a genetic bottleneck by comparing observed heterozygosities with heterozygosities expected under mutation-drift equilibrium with Bottleneck 1.2. If a bottleneck has occurred, there should be a loss of rare alleles, leading to an excess of heterozygotes compared with expectations under mutation-drift equilibrium (Cornuet and Luikart 1996). Because we used microsatellites, alleles that are similar in size are likely to be more closely related than alleles that are more different in size. We thus used the stepwise mutation models (SMM) and two-phase models (TMP) in Bottleneck (Cornuet and Luikart 1996) to generate the predicted heterozygosities. We performed an analysis of population genetic structuring and assignment tests using the model-based clustering analysis implemented in STRUCTURE (Falush et al. 2003; Pritchard et al. 2000). STRUCTURE uses genetic data to assign individuals to a particular number (K) of predefined populations. We performed the STRUCTURE analysis at K = 1 to K = 8 and determined the most likely level of K by determining the modal value of delta K, as described by Evanno et al. (2005). We conducted 100,000 iterations of “burn-in” followed by an additional 10,000 iterations of data collection, using a model of correlated allele frequencies (Falush et al. 2003). In all of the methods using the microsatellite data, the low number of markers (four) we had available could potentially limit our statistical power to detect a bottleneck, and the results should thus be interpreted with caution. However, the markers we used were highly variable, providing a substantial amount of population genetic information, and help us to determine if a bottleneck can be detected using a small number of genetic markers.
Results
The native and introduced ranges differed in mtDNA haplotype composition, based on 56 individuals sampled for eight polymorphic loci (Fig. 1). Seven haplotypes were found in the native range of Australia. Of these, only one (the most common haplotype) also was found in individuals from both quarantine and the introduced range of Florida. The second most common haplotype was found in Florida and quarantine but was absent in Australia. While five of the eight haplotypes were found only in Australia, only two haplotypes were found in Florida, and neither of these was unique to Florida (Fig. 1).

Haplotype network map based on psyllid mtDNA haplotypes. The size of each box corresponds to the number of individuals represented by that haplotype. The individuals are from Australia (A, 22 sampled), Florida (F, 22 sampled) and quarantine (Q, 11 sampled). One line between two haplotypes represents one mutational step between the haplotypes. The small circle is an implied haplotype that was not sampled
Australia shows clear spatial separation of the haplotype groups (Fig. 2). The most common haplotype (A) is found only in the north of the range near Brisbane, which is the area where insects were collected for introduction to Florida. The other haplotypes found in the north of Australia, C and D, differ from haplotype A by only one and two nucleotides, respectively. The only haplotypes found in Florida are A and B; haplotype B differs from haplotype A at a single nucleotide. The A and B haplotypes are intermixed in Florida. The haplotypes found in the central coast of Australia are found only in this region, while the southern range near Sydney contains a different unique haplotype (Fig. 2).

Map of sampling locations and mtDNA haplotypes. Grey dots are the sampling locations. The psyllid population numbers are from Table 1. The letters indicate unique mtDNA haplotypes. The ‘*’ symbol indicates locations where collections were made for the original introductions into Florida
The microsatellite data showed 95 total alleles for the four polymorphic loci used in the study. These four loci were highly polymorphic (16–30 alleles per locus). Gene diversity, allelic diversity, observed and expected heterozygosity, and allelic size range were significantly smaller in the native range of Australia than in the introduced range of Florida (Table 3). The quarantine populations showed the lowest levels of allelic diversity, observed and expected heterozygosity and allelic size range. However, the quarantine populations had higher levels of gene diversity than the native range populations (Table 3). The G–W statistic was 0.277 (±0.009) in the introduced range, which was low, as expected following a bottleneck. However, the G–W statistic in the native range was 0.303 (±0.057) which was not significantly different from the introduced range. Analysis of variance revealed significant effects of source location (Australia, Florida, quarantine) on gene diversity, allelic diversity, observed and expected heterozygosity, and allelic size range, but not the G–W statistic (Table 4).
There was little genetic structure within source locations and low genetic differentiation between the native and introduced ranges for the microsatellite markers. A hierarchal AMOVA showed that only 0.7% of the genetic variation was partitioned among continents, compared with 8.1% among populations and 10.1% among individuals (Table 5). The continents appeared fairly genetically homogenous, with F st = 0.088 in Australia and F st = 0.009 in Florida (Table 6). The quarantine population showed greater structure, with F st = 0.217. The total number of alleles (t1,20 = 2.34, P = 0.03) and allele size range (t1,20 = 2.09, P = 0.05) are larger in Australia than in Florida (Table 6). Observed and expected heterozygosity did not differ between the continents. Genetic diversity (expected heterozygosity) tended to increase from south to north in Australia (Fig. 3).

Genetic diversity cline in Australia. Shown is psyllid genetic diversity (expected heterozygosity) from microsatellite data plotted against population location in Australia. The populations are in order from south to north, corresponding to numbers in Table 1
Comparisons of observed with expected heterozygosity gave no evidence for a genetic bottleneck in the Florida populations. Under the stepwise mutation model (SMM), three loci showed significant heterozygosity deficiency rather than excess (Table 7), which is the opposite of what is expected following a bottleneck. None of the loci showed significant heterozygosity deficiency or excess under the two-phase model (TPM; Table 7). A Wilcoxon test showed that all loci fit mutation-drift equilibrium under the SMM (P = 0.19) and TPM (P = 0.06) models.
Generally more rare and unique (private) alleles appeared in the native range of Australia than in the introduced range of Florida. For example, at loci B6F and B9F, all of the unique alleles were in Australia and none were in Florida. At the 16G locus, nine alleles (two rare and seven private) were found only in Australia, while only four alleles (one rare and three private) were found only in Florida. None of the unique alleles were found in the quarantine populations.
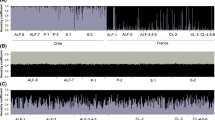
In the STRUCTURE analysis, K = 1 emerged as the most likely number of clusters in the dataset, indicating that both the quarantine populations and the introduced populations were likely drawn from the sampled native (Australian) populations (Fig. 4). Modest population genetic structuring was also evident at K = 2 and K = 3. In particular, some of the quarantine populations (e.g. populations 12, 14 and 15) stood out as relatively uniform, although population 12 (Colony 97-2/4/5) appeared to be distinct from populations 14 and 15 (Colony 98-1 and 99-1, respectively).

a Results of genetic population assignment analysis of microsatellite data using STRUCTURE, at K = 2 and K = 3 and b ΔK values for each level of K. Each individual is represented by a thin vertical bar, and the probability of assignment to each hypothetical populations is indicated by the shaded portion of the bar. The population numbers correspond to those given in Table 1. For convenience, the individuals are shown grouped together by population, and the ordering of individuals within each population is arbitrary
Discussion
The populations of psyllids that we studied went through a known transfer of individuals across continents at a known recent time with a known number of individuals transported to and from specific, identified locations. The populations in Florida have thus passed through a demographic bottleneck of known parameters, giving us a unique opportunity to study the effects of this founder event on the genetics of these populations. We found some evidence of a genetic effect of this demographic bottleneck as a loss of genetic diversity, since there were several microsatellite alleles and mtDNA haplotypes found in the native range but absent in the introduced range. This finding validates theoretical expectations of the genetic consequences of founder events (Nei et al. 1975; Templeton 1980; Barton and Charlesworth 1984). Because a loss of genetic diversity can critically influence population dynamics and evolution, our findings have important implications for the practice of biological control.
The genetic consequences of the demographic bottleneck in the introduced range could also be seen through the genotype and haplotype composition and diversity. In the native range of Australia, there was clear geographic separation of mtDNA haplotypes, with seven total haplotypes found and different sets of haplotypes present in the northern, central, and southern portions of the range. This pattern is consistent with other studies that showed similar genetic differentiation in this same region of the eastern coast of Australia (Arthur et al. 2008). In the introduced range of Florida, there were only two mtDNA haplotypes. One of these haplotypes was found in both the northern range in Australia and in the quarantine population. The other haplotype was found in quarantine and was not in the Australia samples, but was only a single base substitution different from a haplotype that was found in the northern range in Australia. This indicates that the second haplotype was likely present in Australia but at a low enough frequency that it was not present in our sample. Psyllids introduced into Florida through quarantine were originally collected from the northern part of the range in Australia, near Brisbane (Center et al. 2006). Thus only this subset of northern range genotypes were introduced into Florida, indicating a genetic diversity reduction in the introduced range.
Consistent with the results from the mtDNA data, many microsatellite alleles that were found in Australia were absent in Florida, and this was especially true for the unique and less common alleles. This shows that rare alleles are particularly informative and useful in detecting genetic bottlenecks (Slatkin 1985). Only a few alleles were found in Florida and not in Australia. This most likely reflects limited sampling, with these alleles probably occurring at low frequencies in Australia but not in the sample for this study. Alternatively, there could have been mutations at one or more loci in Florida following introduction. Although, mutation rates at microsatellite loci are particularly high (estimated at around 5 × 10−6 for Drosophila in inbred mutation accumulation lines (Vázquez et al. 2000)), it would be unlikely that mutations occurring between the time of the introduction of the psyllids (2002) and the time of our sampling (2004) would have affected our inferences. Thus differences in the genetic composition of the Australia and Florida populations is more likely due to the consequences of the demographic bottleneck than to the occurrence of new mutations.
Interestingly, genetic diversity, averaged across populations, was higher in the introduced range but total genetic diversity within sources was higher in the native range. This finding is consistent with the higher degree of genetic structure and differentiation in Australia than in Florida, which is also as expected following a founder event. The STRUCTURE analysis produced groupings of some individuals consistent with their history and geographic distribution. Overall, the level of clustering with the highest likelihood was observed at K = 1. This result likely reflects the ancestry of the introduced and quarantine populations from ancestral populations in the native range (Australia). Some genetic structure was also observed at higher levels of K. For example, some of the quarantine populations appeared to be relatively homogenous, but distinct from other populations. Individuals in introduced populations were characterized by patterns consistent with a history of admixture, as evidenced by scattered assignment to different populations.
While there was a signature of a bottleneck from the mtDNA data and the population assignment tests using the microsatellite data, other methods using the microsatellite information did not show a bottleneck. These methods included an analysis of genetic structure, comparisons of genetic diversity, the G–W statistic and comparisons of observed with expected heterozygosity. One of the most widely used methods for examining genetic bottlenecks, comparisons of observed with expected heterozygosities under mutation-drift equilibrium (Cornuet and Luikart 1996), showed no evidence for a genetic bottleneck in our study. A two-phase mutation model showed no significant differences between observed and expected heterozygosities, and a stepwise mutation model showed a larger than expected observed heterozygosity at 3 of the 4 loci, in direct contrast with expectations following a genetic bottleneck. The reason these methods did not detect a genetic bottleneck may be that these statistics are strongly influenced by the more common alleles (Hartl and Clark 2007). Genetic bottlenecks may eliminate rare alleles, with little effect on these genetic structure parameters. Thus methods based on examining rare alleles, mtDNA haplotypes, and assignment tests can be more powerful for detecting bottlenecks than those based on genetic structure. It is also important to note that the relatively low number of microsatellite markers used in this study could have limited the power of these methods to detect bottlenecks. However, the results indicate that rare alleles and geographic patterns of mtDNA haplotypes are more informative for detecting bottlenecks than genetic markers with alleles at intermediate frequencies.
The microsatellite data showed low genetic structure, with little differentiation between the native and introduced ranges and a very low amount of the total genetic variation explained by location. The low genetic structure was shown both within populations and among populations based on hierarchial AMOVA. This would indicate that the psyllids are relatively homogeneous genetically both within and across the native and introduced ranges. However, a low value of F ST does not necessarily indicate low differentiation among populations, especially for highly variable loci such as microsatellites (Jost 2008). The lack of spatial genetic structure in the introduced range is not surprising. Because the introduction to Florida occurred recently (in 2002), there likely has been insufficient time for genetic differentiation among populations in the introduced range.
Like our study, several others also showed that recent founder events produced a loss of rare alleles but did not reduce heterozygosity. Hufbauer et al. (2004) examined the population genetics of a parasitoid wasp (Aphidus ervi) introduced as a biological control agent using microsatellite markers and mitochondrial DNA. They found no significant difference in heterozygosity between native and introduced range insects, but they did find substantial genetic structuring and differentiation, with several distinct haplotypes separating the different source locations and a reduction in allelic diversity, especially rare alleles, in the introduced range. Similarly, Baker et al. (2003) examined the genetics of another parasitoid wasp introduced for biological control and also found no loss of heterozygosity but a loss of genetic diversity and rare alleles in the introduced range.
While the founder effect has been well described theoretically (Barton and Charlesworth 1984), the degree to which founding events result in a bottleneck in natural populations as well as the population genetic consequences of species introductions in general is less well understood. Recently, it has also become clear that the genetic consequences of species introductions can be complex. In some cases, introduced populations have lost large amounts of genetic diversity; in other cases, genetic diversity is higher in introduced populations than in native source populations. For example, genetic studies found no evidence of a bottleneck in introduced populations of the Western corn rootworm, Diabrotica virgifera (Kim and Sappington 2005), in the oriental fruit fly, Bactrocera dorsalis (Aketarawong et al. 2007), or in the walnut husk fly, Rhagoletis completa (Chen et al. 2006). Genetic bottlenecks may be uncommon in invasive species because of multiple introductions, or because species that undergo bottlenecks may not establish or become invasive. Furthermore, there is often a lag phase during which the population levels of an introduced species remain low before eventually growing at a rapid rate (Cox 2004). During this latency period, multiple introductions can occur and may offset the loss of genetic diversity in the original introduction. Finally, different introduced species may hybridize with each other or with native species in their new range, resulting in higher levels of genetic diversity relative to source populations in the invader’s native range (Ellstrand and Schierenbeck 2000; Suarez and Tsutsu 2008).
Despite the loss of genetic diversity that has occurred from founder events in introduced species, many of those species have been highly abundant and successful, at least over the short term following introduction. Ross and Shoemaker (2008) found a vast population expansion of the invasive fire ant (Solenopsis invicta) despite a substantial loss of genetic diversity that appeared to be caused by the introduction of a very small number of individuals. Similarly, Argentine ants attain enormous population sizes in their introduced range despite losing substantial genetic diversity during introduction and establishment (Tsutsui et al. 2000; Suarez et al. 2008).
Loss of genetic diversity associated with bottlenecks should be considered in biological control programs. The loss of population fitness associated with a loss of genetic diversity is expected and well known from many studies of rare species (Avise and Hamrick 1996; Frankham and Ballou 2003). Furthermore, a loss of genetic diversity may not be a problem over the short term but may leave the population more susceptible to disease and reduce the ability of the population to evolve following future changes in environmental conditions or in the target species. Thus future biological control programs should consider widespread sampling in the source location, maintaining large populations in quarantine facilities and introducing large numbers of these individuals into the new range, and repeated sampling from the source location and introductions into the new range to limit the loss of genetic diversity in the biological control agent (Madeira et al. 2001).
References
Aketarawong N, Bonizzoni M, Thanaphum S, Gomulski LM, Gasperi G, Malacrida AR, Gugliemino CR (2007) Inferences on the population structure and colonization process of the invasive oriental fruit fly, Bactrocera dorsalis (Hendel). Mol Ecol 16:3522–3532
Arthur AL, Weeks AR, Sgrò CM (2008) Investigating latitudinal clines for life history and stress resistance traits in Drosophila simulans from eastern Australia. J Evol Biol 21:1470–1479
Avise JC, Hamrick JL (1996) Conservation genetics: case histories from nature. Chapman and Hall, New York
Baker DA, Loxdale HD, Edwards OR (2003) Genetic variation and founder effects in the parasitoid wasp, Diaeretiella rapae (M’intosh) (Hymenoptera: Braconidae: Aphidiidae), affecting its potential as a biological control agent. Mol Ecol 12:3303–3311
Barker JSF, Frydenberg J, González J, Davies HI, Ruiz A, Sørensen JG, Loeschcke V (2009) Bottlenecks, population differentiation and apparent selection at microsatellite loci in Australian Drosophila buzzatii. Heredity 102:389–401
Barton NH, Charlesworth B (1984) Genetic revolutions, founder effects and speciation. Annu Rev Ecol Syst 15:133–164
Center TD, Pratt PD, Tipping PW, Rayamajhi MB, Van TK, Wineriter SA, Dray FA, Purcell MF (2006) Field colonization, population growth, and dispersal of Boreioglycaspis melaleucae Moore, a biological control agent of the invasive tree Melaleuca quinquenervia (Cav.) Blake. Biol Control 39:363–374
Chen YH, Opp SB, Berlocher SH, Roderick GK (2006) Are bottlenecks associated with colonization? Genetic diversity and diapause variation of native and introduced Rhagoletis completa populations. Oecologia 149:656–667
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cox GW (2004) Alien species and evolution: the evolutionary ecology of exotic plants, animals, microbes, and interacting native species. Island Press, Washington, DC
Culliney TW (2005) Benefits of classical biological control for managing invasive plants. Crit Rev Plant Sci 24:131–150
Debach P, Rosen D (1991) Biological control by natural enemies. Cambridge University Press, Cambridge
Ellstrand NC, Schierenbeck K (2000) Hybridization as a stimulus for the evolution of invasiveness in plants? Proc Natl Acad Sci USA 97:7043–7050
Elton CS (1958) The ecology of invasions by animals and plants. Methuen, London
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Frankham R, Ballou JD (2003) Introduction to conservation genetics. University of Cambridge, Cambridge, UK
Franks SJ, Kral AM, Pratt PD (2006) Herbivory by introduced insects reduces growth and survival of Melaleuca quinquenervia seedlings. Environ Entomol 35:366–372
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Glenn TC, Schable NA (2005) Isolating microsatellite DNA loci. Methods Enzymol 395:202–222
Goolsby JA, de Barro PJ, Makinson JR, Pemberton RW, Hartley DM, Frohlich DR (2006) Matching the origin of an invasive weed for selection of a herbivore haplotype for a biological control programme. Mol Ecol 15:287–297
Goudet J (1995) Fstat version 1.2: a computer program to calculate Fstatistics. J Hered 86:485–486
Hartl DL, Clark AG (2007) Principles of population genetics, 4th edn. Sinauer, Sunderland, MA
Hufbauer RA, Bogdanowicz SM, Harrison RG (2004) The population genetics of a biological control introduction: mitochondrial DNA and microsatellite variation in native and introduced populations of Aphidus ervi, a parasitoid wasp. Mol Ecol 13:337–348
Hughes S (1998) STR and nucleic acid analysis software. Regents of the University of California, Davis. http://www.vgl.ucdavis.edu/STRand/
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kim KS, Sappington TW (2005) Genetic structuring of Western corn rootworm (Coleoptera: Chrysomelidae) populations in the United States based on microsatellite loci analysis. Environ Entomol 34:494–503
Louda SM, Pemberton RW, Johnson MT, Follett PA (2003) Nontarget effects—the Achilles’ heel of biological control? Retrospective analyses to reduce risk associated with biocontrol introductions. Annu Rev Entomol 48:365–396
Lunt DH, Zhang DX, Szymura JM, Hewitt GM (1996) The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol 5:153–165
Madeira PT, Hale RE, Center TD, Buckingham GR, Wineriter SA, Purcell M (2001) Whether to release Oxyops vitiosa from a second Australian site onto Florida’s Melaleuca? A molecular approach. Biol Control 46:511–528
Morath SU, Pratt PD, Silvers CS, Center TD (2006) Herbivory by Boreioglycaspis melaleucae (Hemiptera : Psyllidae) accelerates foliar senescence and abscission in the invasive tree Melaleuca quinquenervia. Environ Entomol 35:1372–1378
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Pemberton RW (2000) Predictable risk to native plants in weed biological control. Oecologia 125:489–494
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purcell MF, Balciunas JK, Jones P (1997) Biology and host-range of Boreioglycaspis melaleucae (Hemiptera: Psyllidae), potential biological control agent for Melaleuca quinquenervia (Myrtaceae). Environ Entomol 26:366–372
Rayamajhi MB, Pratt PD, Center TD, Tipping PW, Van TK (2008) Aboveground biomass of an invasive tree melaleuca (Melaleuca quinquenervia) before and after herbivory by adventive and introduced natural enemies: a temporal case study in Florida. Weed Sci 56:451–456
Ross KG, Shoemaker DD (2008) Estimation of the number of founders of an invasive pest insect population: the fire ant Solenopsis invicta in the USA. Proc R Soc Lond B Biol Sci 275:2231–2240
Serbesoff-King K (2003) Melaleuca in Florida: a literature review on the taxonomy, distribution, biology, ecology, economic importance and control measures. J Aquat Plant Manag 41:98–112
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39:53–65
Suarez AV, Tsutsui ND (2008) The evolutionary consequences of biological invasions. Mol Ecol 17:351–360
Suarez AV, Holway DA, Tsutsui ND (2008) Genetics and behavior of a colonizing species: the invasive Argentine ant. Am Nat 172:S72–S84
Templeton AR (1980) The theory of speciation via the founder principle. Genetics 94:11–20
Tipping PW, Martin MR, Pratt PD, Center TD, Rayamajhi MB (2008) Suppression of growth and reproduction of an exotic invasive tree by two introduced insects. Biol Control 44:235–241
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proc Natl Acad Sci USA 97:5948–5953
Vázquez JF, Pérez T, Albornoz J, Domínguez A (2000) Estimation of microsatellite mutation rates in Drosophila melanogaster. Genet Res (Camb) 76:323–326
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wineriter SA, Buckingham GR, Franks JH (2003) Host range of Boreioglycaspis melaleucae Moore (Hemiptera: Psyllidae), a potential biocontrol agent of Melaleuca quinquenervia (Cav.) S.T. Blake (Myrtaceae), under quarantine. Biol Control 27:273–292
Acknowledgments
We thank Robyn Chiarelli and Marguerite Stetson for field work and Paul Madeira, Amy Smith and Marguerite Stetson for laboratory work. Matthew Purcell (CSIRO) provided psyllids from Australia. Microsatellites were developed by Travis Glenn and Mandy Schable (University of Georgia). An anonymous reviewer provided helpful comments. This research was funded by a USDA/ARS area-wide management grant awarded to PD Pratt, a grant to SJ Franks from the Fordham University Research Office, and grants from the USDA (NRI-CGP 2004-3502-14865) and the California Structural Pest Control Board to ND Tsutsui.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Franks, S.J., Pratt, P.D. & Tsutsui, N.D. The genetic consequences of a demographic bottleneck in an introduced biological control insect. Conserv Genet 12, 201–211 (2011). https://doi.org/10.1007/s10592-010-0133-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0133-5