
Abstract
Germline mutations affecting the retinoblastoma gene (RB1) predispose to inherited retinoblastomas but also other malignancies, including breast cancer. While somatic RB1 mutations have been detected in different malignancies, information about the potential role of RB1 mutations in breast cancer is limited. Recently, we discovered RB1 mutations to be associated with resistance to anthracyclines/mitomycin in primary breast cancer. The present work is the first report evaluating RB1 mutation and epigenetic status in metastatic breast cancer. Among 148 breast cancer samples analyzed by MLPA, four samples harbored intragenic deletions/duplications: Thus, exons 1–2 were deleted in two tumors and exons 21–23 in one tumor, while one sample harbored duplication of exons 18–23. The entire RB1 gene was duplicated in two tumors and multiple amplifications were revealed in one sample. Reduced copy number was observed in 17 samples (11.5%). No point mutation or promoter hypermethylation was discovered (n = 38 and 114 tumors analyzed, respectively). Interestingly, among seven tumors expressing lack of response to epirubicin, two samples harbored alterations in RB1, contrasting none out of 16 tumors with stable disease or an objective response (P = 0.08). In summary, the frequency of RB1 alterations in metastatic lesions was not increased when compared to primary breast cancer, indicating that RB1 alterations do not play a major role in metastatic development. While a non-significant association suggesting RB1 alterations to be linked to therapy resistance was observed, our data do not suggest a major role for RB1 alterations explaining acquired drug resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The retinoblastoma protein plays a key role regulating cell cycle [1]. RB1 (the gene coding for the retinoblastoma protein) has been found mutated in several types of cancer [2–5], and germline mutations in the RB1 gene lead to retinoblastomas, in general diagnosed within the first year of life [6].
Breast cancer is a heterogeneous category of tumors, characterized by different classes of gene expression profiles [7]. However, while hundreds of genes may be mutated, amplified or epigenetically deregulated, emerging evidence suggests a limited number of gene alterations to be responsible for malignant transformation as well as tumor propagation. Thus, the hypothesis defining cancer gene mutations into “drivers” versus “passengers” is gaining increasing support [8–10]. Based on this hypothesis, mutations affecting a limited number of genes (10–20 only) are considered to be of vital importance to tumor propagation [10, 11]; mutations affecting other genes are considered “passengers”, generated randomly through genetic instability without providing a growth advantage under selection “pressure” in a tumor.
Emerging evidence suggests genes for which germline mutations are associated with cancer risk (e.g., TP53, CDKN2A, and RB1) often to be somatically inactivated in sporadic tumors. While retinoblastoma survivors harboring RB1 mutations are at enhanced risk of developing breast cancer [12], the potential role of somatic RB1 gene defects in breast cancer is poorly understood. While some studies have reported loss of pRb immunostaining [13] or LOH [14] in sporadic breast cancer, little is known with respect to promoter methylation status. In a recent study, we found three point mutations and two intragenetic deletions in a total of 71 locally advanced primary breast cancers [15]. Moreover, three out of a total of four patients carrying such mutations revealed resistance towards anthracycline- or mitomycin-based chemotherapy.
Response rate to chemotherapy is lower in metastatic as compared to primary breast cancer, and metastatic disease inevitably progress towards a state of multidrug resistance [16]. Based on its key role in regulating cellular growth [1] and our recent findings of RB1 mutations associated with drug resistance [15], we postulated mutated pRb protein to be a “driver”, providing a survival advantage for RB1-mutated metastatic breast cancer cells. Thus, the aim of this study was to evaluate the incidence of RB1 genetic and epigenetic disturbances in metastatic breast cancer deposits. In addition, we correlated potential alterations to response to anthracycline containing chemotherapy.
Materials and methods
Patients
All samples were collected from a secondary, metastatic site in patients initially diagnosed with primary breast cancer; this population does not overlap with the primary breast cancer patients enrolled in our previous study [15]. Assembled information on adjuvant treatment following surgery of primary tumor, treatment of metastatic disease prior to biopsy, ER/PgR status, as well as localization of metastatic deposits is provided for the patients in the core cohort for which we performed complete gene sequencing (35 out of 38 with available records) (Table 1) with individual details for those patients harboring RB1 alterations (Table 2).
Snap-frozen tumor samples were collected from a total of 155 patients suffering form metastatic breast cancer. From this cohort, 148 samples were analyzed for gene dosage variation by MLPA, 114 samples for promoter hypermethylation, while 38 samples were screened for mutations throughout the open reading frame using RT-PCR followed by complete sequencing (core cohort). All patients gave written informed consent and the study was approved by the Regional Ethical Committee. While patients received different types of chemotherapy, RB1 status could be correlated to response to subsequent epirubicin monotherapy (applied immediately after biopsy collection) among 23 patients.
Genomic DNA purification
Genomic DNA from tumor tissue was purified using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions.
RNA purification and cDNA synthesis
Total RNA was purified from snap-frozen tumor biopsies using Trizol (Life technologies) extraction and dissolved in DEPC-treated deionised water. cDNA was synthesized by reverse transcription using the Transcriptor reverse transcriptase system (Roche).
Analysis of promoter methylation
Genomic DNAs were subjected to bisulfite conversion using the CpGenome DNA Modification Kit (Intergen) according to the manufacturer’s protocol and screened by promoter methylation- and unmethylation-specific PCRs (MSP and USP).
Both the MSP and USP [17] were performed by an initial 5 min DNA denaturation step at 95°C followed by 35 cycles of 30 s at 94°C, 30 s at 56.6°C and 60 s at 72°C before a final elongation step at 72°C for 5 min. The 50 μl reaction solution for both PCRs consisted of 2.5 U AmpliTaq Gold DNA Polymerase (Applied Biosystems), 1× PCR buffer, 1.5 mM MgCl2, 0.1 mM of each deoxynucleotide triphosphate, 0.2 μM of each primer (Primers are listed in Table 3), and 2 μl of modified genomic DNA.
Multiplex ligation-dependent probe amplification (MLPA)
MLPA analysis of 5 μl genomic DNA from 148 patients was performed using SALSA MLPA RB1 kit (MRC Holland, Amsterdam, The Netherlands) as previously described [15]. The peak areas achieved using RB1 specific probes in each patient sample was first normalized by the average of peak areas achieved by control probes specific for locations different from chromosome 13. A corresponding calculation was performed for two parallels of pooled DNA from 10 healthy individuals. A final ratio was then calculated by dividing the value from the patient samples by the matching value from the pool of healthy individuals. If this ratio was below 0.75, the sample was scored to have reduced copy number; If the ratio was above 1.25 the sample was scored as having increased copy number. In addition, a negative sample (H2O) was included in each run.
PCR amplification of RB1 from cDNA
Amplification of RB1 sequences using cDNA as template was performed by nested PCR. In the first round of PCR, two partly overlapping fragments designated A and B, covering the complete reading frame of RB1, were amplified under the following conditions: 94°C for 5 min, 40 cycles at 94°C for 30 s, 45°C for 30 s and 72°C for 2 min and a final extension at 72°C for 4 min. In the second round of PCR, the A and B fragments were used as templates to amplify the smaller products A1, A2, A3, B1, B2, B3 and B4. The thermocycling conditions for the second round PCRs were an initial step of 5 min denaturation at 94°C followed by 40 cycles of 30 s at 94°C, 30 s at optimized annealing temperature and 60 s (A2, A3, B1, B2, B3, and B4) or 20 s (A1) at 72°C. As a final extension step, samples were incubated for 5 min at 72°C. All primers and annealing temperatures are listed in Table 3. Each PCR was carried out in a 50 μl reaction solution consisting of 0.5 U Dynazyme EXT DNA polymerase, 1× PCR buffer, 5% DMSO, 0.2 mM of each deoxynuclotide triphosphate, 0.2 μM of each primer, and 20 μl cDNA (first round of amplification) or 1 μl of corresponding first-round PCR product (second round of amplification).
PCR amplification of RB1 from genomic DNA
PCR amplification of the RB1 exons 2, 3, 4, 5, 6, 7, 21, and 27 from genomic DNA was carried out using an initial denaturation at 94°C for 5 min, followed by 40 cycles at 94°C for 30 s, optimized annealing temperature (Table 3) for 30 s and 72°C for 30 s. The final elongation was carried out at 72°C for 5 min. The PCR amplification of the exons was performed in a 50 μl reaction solution consisting of 2.5 U AmpliTaq Gold DNA Polymerase (Applied Biosystems), 1× PCR buffer, 1.5 mM MgCl2, 0.1 mM of each deoxynucleotide triphosphate, 0.2 μM of each primer (Table 3), and 1 μl of genomic DNA.
PCR condition for amplification of the RB1 exons 8, 10, 11, 14, 15, 17, 18, and 23 consisted of an initial 5 min denaturation at 94°C, 35 cycles of 94°C for 1 min, optimized annealing temperature (Table 3) for 5 s, and 72°C for 10 s followed by a final step at 72°C for 7 min. The reactions were carried out in a 25 μl solution containing 0.63 U KOD XL DNA polymerase (Novagen), 1× PCR buffer, 0.2 mM of each deoxynucleotide triphosphate, 0.2 μM of each primer (Table 3), and 1 μl of genomic DNA.
TOPO-TA cloning of PCR products
For weak PCR products resulting in non-satisfactory sequence quality, the reaction products were cloned into the pCR 2.1 TOPO vector (Invitrogen) for individual analysis. Resulting colonies were screened by amplifying the insert using M13 PCR (according to the manufacturer’s instructions). Colonies containing insert of correct length were cultured and the corresponding plasmids were purified by Qiagen spin miniprep kit (Qiagen) and sequenced. A minimum of 10 individual clones were sequenced from each sample.
DNA sequencing
DNA sequencing was performed directly on 1–5 μl of the PCR products or plasmids using Big Dye terminator mix (Applied Biosystems) and primers specific for each fragment. An initial denaturation at 94°C for 5 min was followed by 40 cycles of 94°C for 15 s, 50°C for 5 s and 60°C for 4 min. Capillary gel electrophoresis, data collection and sequence analysis were performed using an automated DNA sequencer (ABI 3700, Perkin–Elmer Biosystems), and the resulting sequences were compared with the RB1 wild-type sequence (GenBank accession number L11910).
Statistical analysis
Statistical calculations were performed using the SPSS 15.0 software and http://www.quantitativeskills.com/sisa/. For Fisher exact tests, P-values are given as cumulative and two-sided.
Results
Complete sequencing on cDNA was performed on 38 metastatic tumor deposits (core cohort). For some of the tumor samples, it was not possible to obtain RT-PCR products for a selection of the expected fragments. Thus, we amplified the corresponding exons (2, 3, 4, 5, 6, 7, 21, and 27) from genomic DNA. The complete RB1 reading frame excluding exon 1 was analyzed in 35 samples. As for the three remaining cases, the examination covered the complete reading frame excluding exons 1–5 due to limited tumor DNA in combination with high GC content causing technical problems. Contrasting our previous findings in non-matching primary breast cancer [15], no single point mutation was discovered in any of the metastatic samples.
Due to the potential complication of nonsense mutations and subsequent nonsense-mediated decay of RB1 mRNA, our results were validated by analyzing exons 8, 10, 11, 14, 15, 17, 18 and 23 on genomic DNA from 28 out of 38 patients in the core cohort from whom DNA was available. These exons were analyzed since they have the highest frequencies of nonsense mutations in retinoblastoma patients ([18] and http://rb1-lovd.d-lohmann.de/). In line with our findings using RT-PCR screening, we observed no mutations in these “hot spot” exons when analyzing the same samples using genomic DNA, indicating that the likelihood of any mutation escaping our cDNA sequencing analyses are negligible.
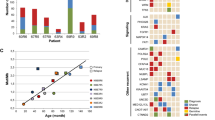
Subsequently, the material was extended and a total of 148 samples were analyzed for larger alterations in RB1 by MLPA. We identified three tumors harboring intragenic deletions (indels) (Table 2). Thus, exons 1–2 were found to be deleted in two tumors (1.4%) and exons 21–23 deleted in one (0.7%) sample (Fig. 1). Surprisingly, we detected RBI duplications in several tumors (Table 2). One tumor harbored a duplication of the exons 18–23, potentially rupturing the full-length pRb protein if located within the reading frame of RB1. Duplication of the entire RB1 locus was observed in two samples (1.4%), while one tumor showed multiple amplifications of the RB1 gene (0.7%). A reduction of the RB1 gene copy number (LOH for the entire locus) was observed in 17 of the samples (11.5%). This was significantly lower than the frequency previously observed by us [15] in non-matching primary tumors (18/71, 25%; P = 0.011).

Schematic presentation of RB1 alterations observed in metastatic breast cancer
Data presented in Tables 1 and 2 reveals that tumors harboring RB1 alterations do not differ from tumors in the core cohort with respect to parameters like pre-biopsy treatment, receptor status and localization of metastatic deposits.
A schematic representation of the RB1 deletions and amplifications observed by MLPA in primary breast tumors examined by us in our previous study [15] and metastatic samples analyzed in this study is presented in Fig. 2.

Diagram showing the distribution of RB1 deletions and amplifications seen by us in metastatic breast cancer lesions analyzed in this study (bottom) and primary breast cancer samples (top) in a previous study [15]
While one of the patients with an exons 1–2 deletion and the patient harboring the exons 21–23 deletion received radiation therapy with non-evaluable response, the second patient harboring exons 1–2 deletion and the tumor containing exons 18–23 duplication both revealed lack of response to epirubicin. Thus, two out of seven tumors expressing resistance to epirubicin harbored alterations in RBI, contrasting none out of 16 tumors with stable disease or an objective response (P = 0.08, including patients analyzed by MLPA and cDNA sequencing with available clinical data on response). Interestingly, combining the data from primary breast cancer [15] and the metastatic samples analyzed in this study, we find 5 out of 24 tumors lacking response to anthracycline or mitomycin therapy to harbor RB1 point mutations or indels contrasting 1 out of 67 with response (P = 0.004).
None of the tumors analyzed revealed promoter hypermethylation (n = 114).
Discussion
Metastatic breast cancer inevitably progress towards multidrug resistance. Based on our recent findings of an association between anthracycline/mitomycin resistance and RB1 mutations in primary breast cancer [15], we hypothesized that RB1 mutations may act as genetic “drivers”, forcing tumor progression and therapy failure. Here, we explored the hypothesis that alterations in RB1 could be enriched in metastatic compared to primary breast cancers.
Contrary to our previous study of RB1 in non-matching primary breast cancer [15], no point mutations were observed in any of the metastatic breast cancer samples analyzed in the present work. However, two different intragenic deletions (indels) were found among three of the 148 metastatic lesions analyzed by MLPA. Our findings are not at conflict with our previous results (three point mutations and two indels in a total of 71 primary cancers), but argue against the hypothesis that RB1 mutations may act as “drivers” to the metastatic process as well as to acquired drug resistance in metastatic breast cancer.
In our previous study of RB1 in primary breast cancer [15], a total of three patients harbored a duplication of the entire RB1 gene. To the best of our knowledge, this was the first time this type of alteration was described for RB1 in breast cancer. As RB1 is a tumor suppressor, one would expect that gene duplication would have an anti-tumorigenic effect. However, the fact that the same phenomenon reappeared together with a case of multiple amplification of the complete RB1 gene (which, to our knowledge has not been described previously for RB1), in non-matching metastatic breast lesions, indicates that duplications at the RB1 locus are not anti-tumorigenic, but rather neutral or potentially tumorigenic.
Although MLPA analysis does not provide information about the exact location of the amplified region, we assume the exons 18–23 duplication to be located intragenic, disrupting the pRb protein function. This duplication was observed in a patient expressing resistance to epirubicin. Given that this alteration could rupture the full-length pRb protein if located within the reading frame of RB1, two tumors (one harboring exons 18–23 duplication and one harboring exons 1–2 deletion) out of a total of seven tumors expressing resistance to epirubicin harbored alterations in RBI, contrasting none out of 16 tumors with stable disease or an objective response (P = 0.08). Although the number of samples are too limited for the data to reach statistical significance, these observations are in line with our previous findings [15] suggesting that alterations in RB1 could contribute to the development of chemotherapy resistance in breast cancer.
Methylation of the RB1 promoter has been observed in various human tumors including one study in breast carcinomas [17, 19, 20]. In the present study, which is the first to analyze for promoter methylation of RB1 in metastatic breast cancer, we found no methylated tumors, indicating that methylation of the promoter region of this gene is not a key event in metastatic breast cancer. This finding is in accordance with what we have previously described for RB1 in locally advanced primary breast cancers [15].
We found the frequency of reduced RB1 gene copy number to be lower among the metastatic lesions as compared to what has been reported by others [14, 21, 22] as well as our own results reporting on non-matching primary breast cancer [15]. This may seem as a paradox, since deletion of a tumor suppressor in theory should provide cells with growth advantages and support tumor progression (“driver”). Our present finding contrasts observations in prostate carcinomas, in which the incidence of RB1 LOH was found higher in metastatic as compared to primary deposits [23]. However, our results are in line with the observations of Ellsworth et al. reporting 25% allelic imbalance for RB1 in primary breast tumor but 17% only in matched axillary lymph node metastases [24]. To the best of our knowledge, we are not aware of any other study reporting RB1 allelic imbalance status in distant metastatic breast cancer lesions. Whether reduced incidence of RB1 allelic imbalance in lymph node as well as distant metastasis indicates clonal selection or presents an acquired phenomenon [25] remains an open issue.
Taken together, we believe our studies of primary [15] and metastatic breast cancer in combination add important information in the quest to understand the role of RB1 alterations in breast cancer behavior. Although not observed in corresponding patients, the finding that the frequency of RB1 alterations did not increase from primary to metastatic lesions argues against a major role for RB1 alterations as drivers during metastatic development. On the other hand, genetic alterations disturbing RB1 function seems to be associated with resistance towards anthracycline therapy. Further studies are warranted to explore the role of other genes involved in the same pathway [26] in anthracycline resistance among primary as well as metastatic breast cancer.
Abbreviations
- LOH:
-
Loss of heterozygosity
- MLPA:
-
Multiplex ligation-dependent probe amplification
- PCR:
-
Polymerase chain reaction
- pRb:
-
Retinoblastoma gene product
- RB1 :
-
Retinoblastoma gene
References
Weinberg RA (1995) The retinoblastoma protein and cell cycle control. Cell 81(3):323–330
Gouyer V, Gazzeri S, Bolon I et al (1998) Mechanism of retinoblastoma gene inactivation in the spectrum of neuroendocrine lung tumors. Am J Respir Cell Mol Biol 18(2):188–196
Kubota Y, Fujinami K, Uemura H et al (1995) Retinoblastoma gene mutations in primary human prostate cancer. Prostate 27(6):314–320
Pinyol M, Bea S, Pla L et al (2007) Inactivation of RB1 in mantle-cell lymphoma detected by nonsense-mediated mRNA decay pathway inhibition and microarray analysis. Blood 109(12):5422–5429
Przybytkowski E, Girouard S, Allard B et al (2003) Widespread bimodal intrachromosomal genomic instability in sporadic breast cancers associated with 13q allelic imbalance. Cancer Res 63(15):4588–4593
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68(4):820–823
Perou CM, Sorlie T, Eisen MB et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Sjoblom T, Jones S, Wood LD et al (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314(5797):268–274
Greenman C, Stephens P, Smith R et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446(7132):153–158
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724
Wood LD, Parsons DW, Jones S et al (2007) The genomic landscapes of human breast and colorectal cancers. Science 318(5853):1108–1113
Marees T, Moll AC, Imhof SM et al (2008) Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst 100(24):1771–1779
Borg A, Zhang QX, Alm P et al (1992) The retinoblastoma gene in breast cancer: allele loss is not correlated with loss of gene protein expression. Cancer Res 52(10):2991–2994
Bieche I, Lidereau R (2000) Loss of heterozygosity at 13q14 correlates with RB1 gene underexpression in human breast cancer. Mol Carcinog 29(3):151–158
Berge EO, Knappskog S, Geisler S et al (2010) Identification and characterization of retinoblastoma gene mutations disturbing apoptosis in human breast cancers. Mol Cancer 9:173
Greenberg PA, Hortobagyi GN, Smith TL et al (1996) Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol 14(8):2197–2205
Simpson DJ, Hibberts NA, McNicol AM et al (2000) Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res 60(5):1211–1216
Valverde JR, Alonso J, Palacios I et al (2005) RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet 6:53
Zemliakova VV, Zhevlova AI, Strel’nikov VV et al (2003) Abnormal methylation of several tumor suppressor genes in sporadic breast cancer. Mol Biol (Mosk) 37(4):696–703
Stirzaker C, Millar DS, Paul CL et al (1997) Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res 57(11):2229–2237
Maitra A, Tavassoli FA, Albores-Saavedra J et al (1999) Molecular abnormalities associated with secretory carcinomas of the breast. Hum Pathol 30(12):1435–1440
Tamura G, Maesawa C, Suzuki Y et al (1994) Improved detection of loss of heterozygosity at retinoblastoma gene locus in human breast carcinoma. Pathol Int 44(1):34–38
Ueda T, Emi M, Suzuki H et al (1999) Identification of a I-cM region of common deletion on 13q14 associated with human prostate cancer. Genes Chromosom Cancer 24(3):183–190
Ellsworth RE, Ellsworth DL, Neatrour DM et al (2005) Allelic imbalance in primary breast carcinomas and metastatic tumors of the axillary lymph nodes. Mol Cancer Res 3(2):71–77
Bissig H, Richter J, Desper R et al (1999) Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am J Pathol 155(1):267–274
Lonning PE (2004) Genes causing inherited cancer as beacons to identify the mechanisms of chemoresistance. Trends Mol Med 10(3):113–118
Acknowledgments
We thank Beryl Leirvaag, Linda Ramsevik and Hildegunn Helle for excellent technical assistance. This work was supported by grants from the Norwegian Cancer Society, Haukeland University Hospital Gene Therapy Program, and Western Norway Regional Health Authority. Parts of this work were performed at Mohn Cancer Research Laboratory, Haukeland University Hospital. The authors declare no competing financial interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Berge, E.O., Knappskog, S., Lillehaug, J.R. et al. Alterations of the retinoblastoma gene in metastatic breast cancer. Clin Exp Metastasis 28, 319–326 (2011). https://doi.org/10.1007/s10585-011-9375-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-011-9375-y