
Abstract
In recent years, our views on how DNA and genes are organised and regulated have evolved significantly. One example is provided by reports that single DNA strands in the double helix could carry distinct forms of information. That chromatids carrying old and nascently replicated DNA strands are recognised by the mitotic machinery, then segregated in a concerted way to distinct daughter cells after cell division is remarkable. Notably, this phenomenon in several cases has been associated with the cell fate choice of resulting daughter cells. Here, we review the evidence for asymmetric or template DNA strand segregation in mammals with a focus on skeletal muscle.
Similar content being viewed by others


Introduction
The seminal experiments by Meselson and Stahl demonstrated the semi-conservative nature of DNA replication (Meselson and Stahl 1958). During replication, each original strand of the double helix serves as a template for the synthesis of a new DNA strand. This configuration then results in two hybrid sister chromatids, each containing a ‘new’ and an ‘old’ DNA strand. Since then, it has been generally assumed that chromatids are randomly distributed between the two daughter cells during mitosis. After several rounds of DNA replication, a cell population then contains DNA molecules of different ages but, for practical purposes, of identical sequences due to the relatively high fidelity of the DNA polymerase complex and nucleotide mismatch editing enzyme machinery.
However, the copied DNA sequences might not be equivalent. Their sequences could diverge because of errors arising during DNA replication itself or editing. Furthermore, DNA damage, errors in repair, or genomic rearrangements can contribute to the infidelity of the process. In addition, genome function is reflected by the primary DNA sequence, but also from its modification and spatial organization. Notably, eukaryotic DNA might be covalently modified via cytosines (hydroxy)methylation. It is wrapped around different histones, which are each subject to a combination of a large variety of posttranslational modifications (acetylation, methylation, phosphorylation, ubiquinitation, sumoylation, etc.). These dynamic epigenetic modifications can influence the genome structure and its transcriptional output. In other words, two DNA molecules with identical primary sequences can be treated differently by the transcriptional machinery within the same nucleus. Notable examples are provided by the inactivation of one X-chromosome in female XX mammals, and the specific monoallelic expression of imprinted genes in mammals.
If single DNA molecules with identical primary sequences were not strictly identical, could these DNA molecules then be treated differently by the mitotic machinery? If so, could sister chromatids be identified and segregated in a non-random manner, depending on the age of their DNA strands?
Experiments in the 1960s with plants and mammalian cells in culture showed that nucleotide analogues (3H-Thymidine) were in some cases not diluted after cell divisions (see Tajbakhsh et al. 2009 and Yennek and Tajbakhsh 2013). These rather unusual observations prompted John Cairns to propose the immortal DNA strand hypothesis (Cairns 1975) according to which dividing adult stem cells transmit the nascent DNA to committed daughter cells through invariant asymmetric cell divisions, whereas the template DNA strands of all chromosomes are retained in the stem cells. In so doing, adult stem cells were thought to avoid the accumulation of replication errors (Cairns 1975) and telomere shortening (Potten et al. 2002; Karpowicz et al. 2005), thereby extending their proliferative potential in the long term while preventing tumour development.
Studies in Schizosaccharomyces pombe have suggested an alternative model, one where regulated gene expression can be mediated by the selective segregation of DNA strands (Klar 1994). Here, expressed and non-expressed alleles would segregate to different daughter cells, thereby promoting effective gene regulation through the selection of chromatids differentially expressing critical loci. The ‘silent sister’ hypothesis (Lansdorp 2007) has also been proposed as an alternative interpretation of template DNA strand cosegregation (TDSS) where epigenetic differences between sister chromatids would drive different gene expression patterns in daughter cells, thereby promoting distinct cell fates. This hypothesis could potentially apply to any asymmetric division, from founder cells during development to stem cells and amplifying progenitors in adult organisms.
The immortal strand hypothesis has received much attention since it was proposed, and its premise is still actively debated (for reviews, see Rando 2007; Lansdorp 2007; Lew et al. 2008; Tajbakhsh and Gonzalez 2009). Some studies have refuted this notion for mammalian hematopoietic and skin stem cells (Kiel et al. 2007; Waghmare et al. 2008; Sotiropoulou et al. 2008), and conflicting results were obtained for the intestinal epithelium (Potten et al. 2002; Falconer et al. 2010; Quyn et al. 2010; Escobar et al. 2011; Steinhauser et al. 2012). Much of the debate arises from (1) uncertainties about the identity or stem cell status of the cells being investigated; (2) uncertainties about the proliferation status (symmetric vs. asymmetric division, cell cycle length, fraction of proliferating vs. quiescent cells, etc. in the population); (3) variabilities in the detection methods used to distinguish the old vs. new chromatids; and (4) variabilities in the time periods used to track the cells under study (long term vs. a few cell divisions). To find out if non-random DNA segregation does occur in a particular tissue, one should ask in which cells, when and which chromosome(s) is (are) involved. Arguably, the most convincing evidence is provided by experiments with mouse skeletal muscle, yet its relationship with cell fate determination, and the mechanism guiding asymmetry of DNA segregation remains unknown. In this review, we will highlight the current knowledge about the physiology of muscle stem cells during development, homeostasis, and tissue repair and highlight differences and similarities in selected systems with a focus on mouse skeletal muscle. We also propose potential mechanisms that might allow asymmetric labelling, recognition, and segregation of sister chromatids.
Skeletal muscle stem cells, growth, and regeneration
Stem cells with regenerative potential in skeletal muscle
Adult skeletal muscle stem cells, or satellite cells, are located between the muscle fibre and the basement membrane ensheathing it. This cell population which is largely quiescent accounts for a low proportion (<5 %) of nuclei in homeostatic adult muscles. Lineage tracing studies have shown that they derive from embryonic Pax7-expressing cells that become lineage restricted at E12.5 (Tajbakhsh 2009; Gayraud-Morel et al. 2009). During regeneration, they exit quiescence, undergo a proliferative expansion phase, and generate myoblasts that differentiate into muscle fibres by fusing with each other, or with nascent or damaged fibres (Reznik 1969; Gayraud-Morel et al. 2009). A fraction that does not differentiate will self-renew and reconstitute a de novo satellite cell pool. During myogenic commitment, Pax7 expression is downregulated. Similarly, satellite cells fuse to existing fibres in growing muscles during development and perinatal growth (Moss and Leblond 1971; White et al. 2010). Different degrees of heterogeneity have been identified among skeletal muscle stem cells, based on embryological origin, association with the fibre, type of cell division, and age of the muscle (Ono et al. 2010; for review, see Tajbakhsh 2009; Biressi and Rando 2010). Other non-satellite cell types with distinct anatomical locations have also been reported to exhibit regenerative capacity after transplantation. These include Pw1+ interstitial cells in postnatal skeletal muscle (Mitchell et al. 2010) and mesoangioblasts (Tedesco and Cossu 2012).
Different surface markers are used to identify and isolate muscle stem cells by FACS (Kuang and Rudnicki 2008; Bosnakovski et al. 2008). The use of a transgenic Tg:Pax7-nGFP mouse (Sambasivan et al. 2009) has circumvented the need to rely on surface markers that can be affected by the enzymes used to dissociate satellite cells from the myofibres. Based on GFP intensity, and therefore endogenous Pax7 expression, satellite cells can be isolated directly by FACS and fractionated into different subpopulations. Interestingly, Pax7-nGFPHi and Pax7-nGFPLo fractions that represent the top or bottom 10 % of the number of cells in the GFP+ satellite cell population exhibit distinct properties, both during quiescence and proliferation (see below and Rocheteau et al. 2012).
Strategies for promoting skeletal muscle degeneration and regeneration
Different methods have been developed to study skeletal muscle regeneration in animal models by inducing partial or complete muscle damage (Schultz et al. 1985; Irintchev and Wernig 1987; Lefaucheur and Sebille 1995; Gayraud-Morel et al. 2009). Myotoxins extracted from snake venoms are the most widely used to induce muscle regeneration. Cardiotoxin is a protein kinase C inhibitor that induces depolarization and contraction of muscle cells, membrane disorganization, and cell lysis. Notexin is a phospholipase A2 that blocks neuromuscular transmission by inhibiting acetylcholine release. Other methods include injection of barium chloride, crushing or freezing, denervation/devascularisation, necrosis by transplantation of the muscle, and intensive exercise (Gayraud-Morel et al. 2009). Muscle injury is followed by phases of degeneration and necrosis, then regeneration involving inflammation, vasculogenesis, and myofibre reconstruction via satellite cells. However, the kinetics and amplitude of each phase vary depending on the muscle, the nature and the extent of the injury. Lineage progression from stem to differentiated cells can be monitored by the successive expression of markers of activation (e.g. Myod, CyclinA, Ki67, PCNA), commitment (e.g. Desmin), and differentiation (e.g. Myogenin; Kuang and Rudnicki 2008).
Investigating non-random DNA segregation in muscle
Methods for selective DNA strand labelling with nucleotide analogues
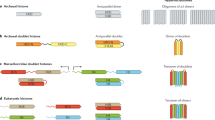
Experimental approaches involve pulse-labelling of template and/or nascent DNA strands by administration of thymidine analogues [5-bromo-2′-deoxyuridine (BrdU), 5-chloro-2′-deoxyuridine (CldU), 5-ethynyl-2′-deoxyuridine (EdU), 5-iodo-2′-deoxyuridine (IdU)] and observation of their distribution over subsequent cell divisions during the chase period, together with their association with cell fate markers. It is important to consider the different labelling regimes used to assess TDSS, namely inclusion (old DNA strand labelled) and exclusion (new DNA strand labelled; Fig. 1). During the following discussion, we will refer to the cell division allowing readout of TDSS as ‘T2’, the previous divisions as ‘T1’ and ‘T0’ (see Fig. 1 for details). Using these protocols, several critical parameters should be met for observing TDSS: (1) selective labelling of template vs. nascent DNA; (2) chase period corresponding to a predetermined cell cycle time to assess T1 and T2 empirically; (3) rigorous methods of analysis to identify non-random DNA segregation and true daughter cell identities.

Tracking TDSS via inclusion or exclusion labelling protocols T2 is referred as the division at which TDSS is observed; T1 and T0 as the preceding divisions. a Inclusion protocol. Template DNA is labelled over multiple cell divisions with a first thymidine analogue until T0. During exponential cell expansion, it is assumed that cells containing the initial oldest DNA strands (unlabelled) are diluted in the population among cells that undergo successive rounds of divisions, thereby resulting in both DNA strands being labelled. Cells are then allowed to complete two divisions (T1 and T2) during the chase period, with a second thymidine analogue (to check the proliferation status of cells labelled with the 1st analogue). In case of TDSS, the daughter cell that inherits the oldest (newest, respectively) DNA strands is positive (negative respectively) for the first DNA label, and both daughter cells are positive for the second DNA label. b Exclusion protocol. Nascent DNA is labelled over one cell division with a first label, then cells undergo mitosis (T1). Cells that perform a division during the chase (in the presence of a second analogue to monitor continued proliferation) give rise to T2 cells. In case of TDSS, the daughter cell that inherits the oldest (newest respectively) DNA strands is negative (positive respectively) for the first DNA label, and both daughter cells are positive for the second DNA label
Proliferation state of the cells exiting dormancy or quiescence
As indicated above, an important parameter is the proliferation status of the cell population of interest. Several studies involve the activation of a quiescent stem cell population. This raises the following questions: Are some, or all, cells activated? How rapidly does this take place? How often do the cells divide subsequently? Once activated, this population could perform symmetric (self-renewal or differentiation type), asymmetric (self-renewal/differentiation or commitment/differentiation type) divisions, or both. In mouse skeletal muscle, we characterised the Pax7-expressing population, using transgenic Tg:Pax7-nGFP mice (Rocheteau et al. 2012). Although both Pax7-nGFPHi and Pax7-nGFPLo subpopulations are quiescent in adult muscle, they exhibit heterogeneous properties with distinct metabolic activities (lower in Pax7-nGFPHi than in the Pax7-nGFPLo) and gene expression patterns (stem cell markers Cxcr4, Cd34, Pax7: GFPHi > GFPLo; commitment markers Sca1, Desmin, Myog: GFPHi < GFPLo). Furthermore, the quiescent Pax7-nGFPHi subpopulation takes longer than the quiescent Pax7-nGFPLo cells to activate and enter the cell cycle in vitro (first division within 33 vs. 26 h, respectively) and in vivo (S-phase entry within 24 vs. 18 h, respectively). Therefore, Pax7-nGFPHi cells are in a more ‘dormant’ cell state than the quiescent Pax7-nGFPLo cells. Subsequently, each population divides every 7–8 h, in vitro in low oxygen (3–5 %) as in vivo.
Empirical detection of non-random DNA segregation patterns
Different labelling protocols are used to assess TDSS, with single or double thymidine analogues. Although in vivo doses employed are comparable between different studies, the number and timing of injections differ. These doses need to be relatively low, given the toxicity of these halogenated analogues, and their ability to affect gene expression, for example, repression of Myod (Ogino et al. 2002) and cell differentiation (Bischoff and Holtzer 1970). At lower doses of BrdU Pax7 and Myod expression, clonogenicity, proliferation, and differentiation potential were not affected (Shinin et al. 2006). When discriminating between DNA-label+ and DNA-label− cells after a chase period, the sensitivity of detection might vary according to the dose of analogue used and the detection protocol (BrdU, CldU, and IdU detected by immunochemistry, EdU by click-chemistry).
As indicated above, two cell divisions are required to observe TDSS empirically. As far as in vivo chase divisions are concerned, it is important to determine the clearance time of the thymidine analogues (∼8 h in vivo; unpublished observation). After DNA labelling of proliferating muscle stem cells, two different methods are employed to demonstrate TDSS. With both protocols, a mother cell performing TDSS should be first-label+ by T1 (cf Fig. 1), and give rise to two daughter cells, one first-label+ and one first-label− at T2. This can be assessed at the single-cell level or at the population level. Clonal cell analysis is the most powerful method currently in use, although evidence for clonality should be rigorously demonstrated as cells can migrate extensively in vitro as shown by live videomicroscopy (Shinin et al. 2006). Interestingly, one study co-cultured primary myoblasts from WT and fluorescently marked mice (ubiquitous fluorescent Tomato protein, added exogenously), to identify false (Tomato + and −) and true (Tomato-negative) cell pairs (Liu et al. 2012). As cells in contact may not necessarily be true sister cells, unambiguous identification of sister cells also involves the demonstration of a physical connection between daughter cells (e.g. Aurora B kinase immunostaining at the midbody (Terada et al. 1998)). Drugs such as nocodazole or cytochalasin D can be used to prevent cytokinesis between sister cells (Karpowicz et al. 2005; Conboy et al. 2007; Huh et al. 2011; Huh and Sherley 2011), although these might also interfere to some extent with the mechanism of TDSS.
Another important control requires demonstration that mother cells are indeed positive for the first DNA label at T1 (cell division preceeding TDSS readout) because label-negative cells, an outcome of TDSS, might also result from failure to have incorporated the first nucleotide analogue. This is also crucial when analysing TDSS at the population level. For example, by isolating Pax7-nGFPHi and Pax7-nGFPLo populations from regenerating muscle subjected to successive EdU (first label) and BrdU (second label) injections, both subpopulations of myogenic cells were shown to be >95 % positive for both labels in an aliquot taken at T1 (Rocheteau et al. 2012). Label exclusion was then noted in Pax7-nGFPHi but not Pax7-nGFPLo cells after the second division. Conversely, Conboy et al. (2007) applied an exclusion labelling protocol using sequential injections of CldU and IdU to proliferating adult muscle stem cells, and observed the asymmetric distribution of CldU in cell pairs at T2. Therefore, analyses of TDSS should be carried out at the single-cell level from homogeneously labelled populations.
An important distinction between the inclusion (template) and exclusion (nascent) protocols for labelling strands is the persistence of the labelled DNA strands. For labelling of the older template strands, two scenarios are possible: (1) DNA strands that are labelled during symmetric cell divisions, for example during exponential cell expansion, can then act as template strands in subsequent cycles. In this case, original unlabelled template strands will be diluted in the expanding cell population (see Fig. 1a); (2) nascent DNA strands from asymmetric divisions might act as future template strands that will segregate asymmetrically. In either case, two rounds of asymmetric DNA segregation during the chase period would result in labelled template strands and unlabelled nascent strands in daughter cell pairs. If cells divide symmetrically (random segregation) during the chase period, label dilution will compromise the detection of asymmetric segregation patterns (six to eight divisions dilutes label to below robust threshold levels for detection). Notably, if template strands are labelled, label retention could in principle persist for successive divisions, if invariant asymmetric divisions follow. Using the exclusion protocol, however, if the labelled nascent DNA strands will segregate randomly, each division will result in retention of half the label, thereby resulting in less than ten labelled chromatids in the cell after two to three divisions (for mouse, 40 chromosomes). A random segregation of these chromatids that is skewed by chance could then be falsely interpreted as a non-random segregation pattern.
Non-random DNA segregation: one, some, or all chromatids?
If protection of the genome from accumulated mutations, and consequently ‘immortal’ DNA strand segregation were the primary mechanism driving this asymmetry, one would expect that all old template strands should segregate asymmetrically. However, reports of TDSS implicating one or a few chromosomes suggest that this might not be the case—therefore, alternative explanations were proposed for this phenomenon. For example, in mouse ES cells, chromosome 7, but not chromosome 11, was initially reported to segregate non-randomly (Armakolas and Klar 2006), yet more recent results by Strand-seq (Falconer et al. 2012) and Chromosome orientation fluorescence in situ hybridization (CO-FISH; Sauer and Klar, this issue) indicate that this is not the case. Non-random segregation of some but not all chromatids was suggested for cells in the mouse intestinal crypt (Falconer et al. 2010), although this study did not discriminate between some or all cells performing TDSS for a subset of chromosomes. Nevertheless, these observations underscore the importance of following the fate of single chromatids by CO-FISH (Bailey et al. 1996; Falconer et al. 2010) or other methods (Falconer et al. 2012).
Non-random DNA segregation is associated with cell fate in skeletal muscle stem cells
Evidence for non-random DNA segregation
In vivo investigation of TDSS in mouse adult skeletal muscle has involved different approaches. In one case, DNA was labelled with BrdU during postnatal growth (P3-P10), followed by a chase period from 1 to 10 weeks (Shinin et al. 2006). Clonal analysis of label-retention in in vitro reactivated adult satellite cells revealed that up to 7 % of the clones showed a single BrdU+ cell, expressing markers of activation (Myod, CyclinA; Fig. 2a).

Evidence for biased DNA segregation in muscle stem cells TDSS was associated (a, b, c) or not (d) with an asymmetric cell fate. a An inclusion protocol was applied to muscle skeletal satellite cells during post-natal growth (post-natal days 3 to 10), followed by a chase in vivo and clonal analysis of label retention in in vitro reactivated adult satellite cells. Up to 7 % of the clones showed a single label-retaining cell, expressing markers of activation (Myod, CyclinA) and stemness (Pax7, Myf5). Additional analysis provided evidence for TDSS in in vivo pulsed/chased daughter cell pairs on isolated muscle fibers (≈7 %) and in vitro (≈1.5 %). TDSS was also directly observed by tracking daughter cells by videomicroscopy (not shown here) (Shinin et al. 2006). b An inclusion protocol was applied to adult Tg:Pax7-nGFP mice where satellite cells were reactivated following notexin injury, then GFPHi and GFPLo satellite cells were sorted. At T1, both subpopulations were homogeneously positive for both DNA labels. At T2, cells negative for the first label were observed in the Pax7-nGFPHi subpopulation, up to 40 %, indicating up to 80 % of cells performing TDSS. Myogenin expression was detected in cells having inherited new DNA strands. In addition, CO-FISH analysis showed that virtually all chromatids were segregated by TDSS. Finally, Pax7-nGFPHi and Pax7-nGFPLo subpopulations expressed (RT-qPCR) different levels of lineage progression markers (Cxcr4, Cd34, Pax7: GFPHi > GFPLo; Sca1, Desmin, Myog: GFPHi < GFPLo), either in resting or regenerating (5 days post-injury) muscle (not shown here; Rocheteau et al. 2012). Tc measured cell cycle time. c An exclusion protocol was applied to adult satellite cells that were reactivated following diffuse cardiotoxin injury. At T1, myogenic progenitors were dissociated from the muscle fibres and allowed to complete the last cell cycle until telophase, producing attached daughter cell pairs; 45 % of cell pairs showed asymmetric distribution of the first label. This was associated with mostly asymmetric expression of Desmin in the label-inheriting cell. Additionally, most cell pairs also expressed Sca1 (referred here to a more upstream marker) in Desmin-negative cell (Conboy et al. 2007). d Exclusion protocols were applied to cKit+ hCardSCs, either to expand hCardSCs clones (Exp. #1) or single cells (Exp. #2). Different chase periods were used. At T2, 5 % of expanding clones showed DNA-labelling positivity in 50 % of the cells. Similarly, 5 % of the single cells gave rise to clones showing one cell positive for the first label, two cells positive for the second label, and the remaining cells negative for both labels. Such clones were referred to as ‘Asymmetric Chromatid Segregation (ACS)’ clones, as opposed to ‘Symmetric Chromatid Segregation (SCS)’ clones where the label was diluted. ACS and SCS clones expressed no lineage commitment markers and showed uniform α-adaptin staining, a reporter of asymmetric cell fate. ACS clones proliferated faster than SCS clones, had longer telomeres, and less expression of senescence or apoptosis markers. The frequency of ACS clones was shown to decrease with the age of donor patients, and upon loss-of-function of Dynein. Finally, ACS clones showed better reconstitution of infarcted hearts than SCS clones (Kajstura et al. 2012). Tc measured cell cycle time
The occurrence of TDSS was also investigated in muscle regeneration paradigms. Adult satellite cells were reactivated by myotoxin injection, serially labelled with thymidine analogues, and examined for DNA strand segregation and expression of cell fate markers (Conboy et al. 2007; Rocheteau et al. 2012; see Fig. 2 for detailed protocols). The myogenic population was fully (Rocheteau et al. 2012) or partially (Conboy et al. 2007) labelled by the first thymidine analogue. The proliferation during the chase period was confirmed by the presence of a second thymidine analogue. Briefly, TDSS was identified in both studies, up to 50 % of the total satellite cell population (Conboy et al. 2007) and 80 % of the Pax7-nGFPHi population (8 % of total; Fig. 2b, c; Rocheteau et al. 2012). It is currently not clear why the frequencies vary between these two studies; it does not appear to result from the labelling regimes as alternative ones (inclusion vs. exclusion, various thymidine analogues) gave similar results.
TDSS has also been investigated in mouse skeletal muscle stem cells and human cardiac stem cells in vitro. When pulse/chase studies were performed in vitro from P3 with skeletal myogenic cells, 1.5 % of cell pairs displayed asymmetric BrdU labelling (Shinin et al. 2006). Similarly, less than 5 % of DNA-labelled asymmetric cell pairs were observed with established myoblast cultures (Conboy et al. 2007). The differences in frequencies from the two studies indicated above and the lower frequencies obtained in vitro underscore the importance of the microenvironment, or niche. Given these observations, it would be interesting to determine if all myogenic cells have the potential to perform TDSS. This raises another issue concerning empirical sampling of frequencies of symmetry and asymmetry. It is currently not clear if all myogenic cells explore both modes of division, perhaps depending on their position within a polarized niche, or whether obligate modes of symmetry or asymmetry define myogenic cell divisions. Isolation of cells at any timepoint provides a snapshot of current events, but not past or future divisions of the cells being sampled. An important consideration is that in the protocol used by Rocheteau and colleagues, virtually all muscle stem cells from injured Tibialis anterior muscle entered the cell cycle within 44 h after injury, and analysis was done 5 days post-injury. Conversely, the protocol used by Conboy and colleagues resulted in a low percentage of CldU-incorporating cells 48 h post-injury, at which point TDSS was examined. This low percentage of CldU incorporation indicates (1) an activation of a subpopulation of muscle stem cells and/or (2) that analyses were performed when muscle stem cells were undergoing their first cell cycles. To resolve these differences and better understand muscle stem cell properties, the dynamics of symmetric vs. asymmetric DNA segregation within activated muscle stem cells at timed intervals following muscle injury will need to be done.
In striking contrast to observations suggesting that the in vivo niche is necessary to maintain TDSS in skeletal muscle, recent work with human cardiac stem cells suggests that this might not always be the case (Kajstura et al. 2012). Clonal analysis of c-Kit+ human cardiac cells labelled in vitro with BrdU identified 7 % of clones with a single BrdU+ cell, the progeny of which were reported to be BrdU−. Notably, those BrdU+ cells within the clones were cycling. In this model, the frequency of non-random segregation remained constant, even after multiple passages. Another intriguing observation is that these cardiac cells in vitro appeared to be in an obligate or invariant mode of asymmetric DNA segregation. Indeed, application of an exclusion protocol (nascent DNA labelled) using CldU and IdU with a chase period extending from one to multiple cell divisions identified 5 % of clones containing only a single CldU+ cell, two IdU+ cells, and the remaining cells negative for both labels (Fig. 2d). In addition, labelling of clones for one cell cycle during their expansionary phase of growth, followed by a single round of chase resulted in label retention in half of the cells in the clone, and exclusion in the remaining cells. These observations suggest that a mother cell dividing by asymmetric DNA segregation gives rise to daughter cells also dividing by TDSS. This intriguing observation that cardiac myogenic cells can perform TDSS in vitro independent of the niche needs to be confirmed in vivo, and should also include the demonstration that all chromatids are involved in this asymmetry.
In summary, parameters such as the cell cycle length, labelling regimes, and rigorous methods of analysis need to be established to observe TDSS. Of note, each of the methods includes an in vitro step to reveal TDSS. Therefore, in vivo assays for TDSS in muscle stem cells need to be developed. This is particularly challenging given the technical difficulties in detecting mitotic muscle stem cells in sufficient numbers (unpublished observations). Nevertheless, Rocheteau and colleagues showed in one series of experiments by CO-FISH that cells isolated directly from the muscle and blocked in mitosis performed TDSS. In addition, live observation of the phenomenon is rendered difficult due to the lack of fluorescent nucleotide analogues with sufficient resolving power. Thus far, only one study has demonstrated the occurrence of TDSS with single chromosome resolution, by CO-FISH (Rocheteau et al. 2012). The mechanisms and function of TDSS in muscle stem cells remain unknown, although the association of this phenomenon with cell fate has been established.
Non-random DNA segregation and cell fate decisions
The silent sister hypothesis presumes that asymmetric DNA segregation could be associated with differential gene expression patterns between daughter cells, possibly driving asymmetric cell fate (Lansdorp 2007). Accordingly, reports of TDSS in skeletal muscle stem cells associated DNA asymmetry with cell fate (Shinin et al. 2006; Conboy et al. 2007; Rocheteau et al. 2012). Clonal analysis of label-retaining muscle stem cells (3 weeks chase in vivo, 6 days in culture) showed a retention of old DNA strands in the less differentiated cells, i.e. those expressing Pax7 (Shinin et al. 2006). Similarly, template DNA strands were inherited by the less differentiated daughter cells during muscle regeneration, based on Pax7 (Rocheteau et al. 2012) or Sca1 expression (Conboy et al. 2007), whilst newly synthesized DNA strands were transmitted to the more committed daughter cells, expressing the differentiation marker Myogenin (Rocheteau et al. 2012) or Desmin (Conboy et al. 2007). We note that some discrepancies exist in the association of some markers with the upstream or downstream cell fates of muscle stem cells in these studies. For example, Sca1 protein was used as an upstream marker by Conboy and colleagues, whereas Rocheteau colleagues showed Sca1 transcripts were more expressed in committed cells (Pax7-GFPLo subpopulation). These differences need to be reconciled. Whether TDSS can be systematically uncoupled from this cell fate decision in skeletal muscle stem cells remains an open question.
In contrast to skeletal myogenic cells, TDSS in cultured human cardiac myogenic cells was not associated with a cell fate decision (Kajstura et al. 2012). The origin and nature of these cells are not yet clear, and there is active debate regarding the presence of tissue-specific stem cells in the heart (Harvey and Tajbakhsh 2012). More studies with other tissues are required to determine to what extent TDSS is associated with a cell fate decision.
Mechanisms that could promote non-random DNA segregation
Non-random DNA segregation has been investigated in a number of systems, from bacteria to a variety of mammalian cells (for review, see Yennek and Tajbakhsh 2013). Mechanisms promoting TDSS remain largely unknown in mammals, but several hypotheses have been proposed (Klar 1994; Lansdorp 2007; Lew et al. 2008; Tajbakhsh and Gonzalez 2009). One can distinguish different levels of regulation involving extracellular signals, cytoplasmic determinants, and chromosomal determinants.
As discussed earlier, the frequency of TDSS in adult skeletal muscle stem cells decreases as they are dissociated from their niche. In addition, we raised the possibility that muscle stem cells might switch between asymmetric and symmetric modes of cell division and DNA segregation during growth and tissue regeneration. As this has not been tested, the possibility should be considered. Therefore, non-random DNA segregation might respond to extracellular cues originating from the niche. These signals could then be transduced by membrane-associated proteins or cytoplasmic determinants, allowing the asymmetric recognition and segregation by the mitotic machinery of ‘marked’ sister chromatids. Sufficient information for non-random segregation of sister chromatids might be present within centromeres, which provide a physical link between DNA and the mitotic apparatus (Fig. 3). This notion needs to be tested empirically. A key point is the establishment of an asymmetry between sister chromatids, beginning during S phase and subsequently maintained, most probably at centromeres.

Possible mechanisms promoting TDSS during cell cycle progression (a) Representation of a mother chromatid in G1, with centromeres associated with ‘old’ nucleosomes and two genes, A and B, of unknown expression status. ‘A’ would be a master regulatory gene, ‘B’ an irrelevant gene for cell fate decisions (e.g. housekeeping). b1 Preferential replication fork progression across the mother centromere can result in asymmetric loading of PCNA on leading and lagging strands. b2 Alternatively, after centromere replication, sister chromatids might be loaded with equal amounts of limiting specific factors within centromeres. Transient pairing of centromeres could then result in the redistribution of such factors on one centromere vs. the other, similar to what is assumed to happen during symmetry breaking between the two X chromosomes in the X-chromosome inactivation model (Navarro and Avner 2010; Augui et al. 2011). c1–4 Primary symmetry breaking according to the asymmetric replication fork progression model or the transient sister chromatid pairing model can result in secondary asymmetries. These can be asymmetric transcription (c1), asymmetric DNA (hydroxy)methylation (c2), asymmetric histone modifications (c3), or asymmetric distribution of ‘new’ and ‘old’ histone pools (c4), within centromeres. d Centromere asymmetry might enable future asymmetric expression of master regulatory genes (‘A’), without affecting ‘housekeeping’ genes (‘B’), according to the silent sister hypothesis (Lansdorp 2007). e Asymmetrically labelled centromeres might be differentially recognized by kinetochore proteins. A crosstalk might exist between centromeres and centrosomes, via cargo proteins shuffling between kinetochores and centrosomes. This could enable the biased segregation of ‘old’ vs. ‘new’ chromatids. f Asymmetric cell fate decision in daughter cells inheriting ‘old’ or ‘new’ sister chromatids
Symmetry breaking between sister chromatid centromeres
Replication, transcription, repair, and maintaining quality control in stem cells
The movement of the replication fork inherently creates an asymmetry between the two template DNA strands, one being replicated by the leading strand apparatus, the other by the lagging strand apparatus. In Escherichia coli, leading and lagging DNA strands can be segregated to different locations in response to the direction of DNA replication (White et al. 2008). Some reports indicate that the speed of progression of the replication fork can vary between the different sides of the origin (Raghuraman et al. 2001; Karnani et al. 2007). In addition, preferential direction of replication can result from a non-random distribution of origins and termination signals (e.g. preferentially on one side of origins), or from the presence of unidirectional terminators, allowing fork progression in a single direction (Rothstein et al. 2000). An elegant example of unidirectional DNA replication is provided by mating type switching in the fission yeast. S. pombe cells exist as two mating types, P and M, that switch during cell divisions. It was shown that unidirectional DNA replication of the mat1 locus directs the imprinting of one of the two sister chromatids during the following round of DNA replication, leading to mating-type switching by recombination (Dalgaard and Klar 1999). The nature of this imprint has been proposed to be a site- and strand-specific break (Arcangioli 1998) or an RNA residue from an Okasaki fragment (Klar 1987; Nielsen and Egel 1989).
It has been proposed that proliferating cell nuclear antigen (PCNA) remains associated with double-strand DNA before removal by the Replication Factor-C in Saccharomyces cerevisiae (Bylund and Burgers 2005). In this case, leading and lagging strands could retain different amounts of PCNA (Fig. 3b1; Lew et al. 2008; Tajbakhsh and Gonzalez 2009). Furthermore, PCNA has been suggested to link DNA replication and inheritance of epigenetic states via CAF-1 protein complex that deposits histone H3-H4 proteins onto replicating DNA (Shibahara and Stillman 1999). As a consequence, biased orientation of replication might induce secondary asymmetry between sister chromatids, for example, in the form of asymmetry in the methylation patterns, histone distribution, or histone modifications. Strikingly, a mutation that inhibits CAF-1-mediated nucleosome formation results in failure to form bilateral asymmetry in the Caenorhabditis elegans nervous system, thereby providing a direct link between left–right asymmetric epigenetic regulation post-replication and bilateral asymmetric cell fates (Nakano et al. 2011).
In addition, strand-specific transcription could determine an asymmetry between sister chromatids, either by itself (Fig. 3c1) or by impinging on the progression of the replication machinery. Strand-specific transcription has been shown to occur at centromeres (Kanellopoulou et al. 2005; Murchison et al. 2005), and to be necessary for the proper organization of pericentric heterochromatin during early mouse development (Probst et al. 2010).
In some cases, robust transcription and DNA replication can occur in opposite directions during S phase. This can result in paused replication forks as opposing tasks compete for function on the same DNA strand. If readthrough at the fork occurs, this could potentially modify the epigenetic signature of the DNA and consequently gene expression (Bermejo et al. 2012).
Other structural configurations that could potentially result in strand-specific asymmetry during replication involves G-quadruplexes. These sequences are four-stranded nucleic acid structures that are associated with many metazoan replication origins (Méchali et al. 2013). They exhibit asymmetrical behaviour during replication, exclude nucleosomes, and their processing is critical for maintaining genome stability (Méchali et al. 2013). We propose that their behaviour during leading and lagging strand synthesis could potentially promote asymmetric behaviours of the replicated DNA. Furthermore, emerging studies point to active and silenced origins that constitute a replicon, and these regions are enriched in epigenetic marks (Méchali et al. 2013). The resetting of these marks could also impact on gene expression, thus providing another potential link between the replication process and gene regulation.
In extreme cases, or during stress, the replisome can remain associated with the DNA fork, resulting in a stalled fork, and this is protected by an inter-S-phase checkpoint (Mizuno et al. 2013). Fork collapse resulting from dissociation of polymerases, or broken forks, can be accommodated if replication restarts by homologous recombination or microhomology-primed re-initiation (Bermejo et al. 2012). It is not known how these processes affect tissue-specific stem cells or their mode of cell division.
In this context, it is worthwhile considering the possibility that stem cells might avoid replication or other types of errors, or preserve their proliferative capacity by limiting telomere shortening. Independently of the immortal strand hypothesis, the notion that adult stem cells might avoid accumulating mutations after DNA replication has not been empirically tested. This is a challenging experiment to do as one should compare cell populations and their progeny for extended periods of time. In addition, the extent of quality control following excision/repair, sister chromatid exchange, or double-stranded break repair has not been evaluated extensively in stem cells. Whether these events impact on or promote TDSS remains an open question (see Lansdorp 2007; Tajbakhsh and Gonzalez 2009, Yennek and Tajbakhsh 2013).
Interestingly, recent findings have pointed to robust repair after DNA damage in stem cells compared to committed cells (Sotiropoulou et al. 2010; Blanpain et al. 2011, S. Tajbakhsh and M. Ricchetti, unpublished results). DNA damage comes in different forms, and repair generally takes place with high fidelity, depending on the choice of the polymerase recruited and its proof-reading capacity (Kunkel 2004; Blanpain et al. 2011). In some cases, however, specific translesion synthesis polymerases copy past DNA lesions that can block replication, and these can exhibit high error rates. Some of these polymerases (e.g. Pol ζ, Pol η, Pol ι, and Pol κ) lack proofreading activity, and introduce errors ranging from 10–1 to 10–3 (Kunkel 2004). Another consideration regarding DNA replication itself is that lagging strands can accumulate less errors, at least in bacterial chromosomal DNA, compared to leading strands (Fijalkowska et al. 1998).
Therefore, defects in processes that determine DNA replication fidelity can confer mutator phenotypes. Although managing errors by selective DNA strand segregation was initially the premise put forth for the immortal strand hypothesis (Cairns 1975, 2006), non-random DNA segregation might result from asymmetries inherent in the replication process itself, or the quality control associated with post-replication events. Although it is estimated that approximately one error occurs during DNA replication for each 109–1010 nucleotides polymerized (Prindle et al. 2010), this has not been measured directly in adult vertebrate stem cells and their progeny over extended periods. Future studies should consider how these different processes, as well as the fidelity of DNA repair following induced DNA damage, contribute to mutation load in tissue-specific cells in long living metazoans.
Thus, quality control is a process that is intimately linked to DNA replication. The recent findings that DNA repair enzymes are associated with the transcriptional machinery and that they can affect gene expression provide a tantilizing link between the maintenance of DNA fidelity and the regulation of gene expression (Compe and Egly 2012). In this context, we note that the tumour suppressor gene p53 was reported to regulate symmetric versus asymmetric cell divisions in cultured cells, where retention of template DNA strands by non-random chromosome segregation was suppressed in p53 null cells (Rambhatla et al. 2005). Given the reported link between p53 and the cell cycle determinant Numb, which can regulate asymmetric cell divisions (Cicalese et al. 2009), this raises the possibility that DNA damage response mediated by p53 or other effectors could link the DNA repair pathway with non-random DNA segregation. We note that TDSS was not fully suppressed in cultured p53 null skeletal muscle stem cells (Shinin et al. 2006). With the higher resolution protocols recently described for prospectively isolated muscle stem cells (Rocheteau et al. 2012), the role of the DNA damage response pathway, as well as p53, could be investigated in more detail.
Non-random DNA segregation and X-chromosome inactivation
In mammalian cells, symmetry breaking between identical DNA sequences occurs during X-chromosome inactivation. It is thus tempting to consider the possibility that the mechanisms involved in X-chromosome inactivation could apply to symmetry breaking between sister chromatids of autosomes. In the inner cell mass of female mouse embryos as well as in female mouse ES cells, one of the two X chromosomes is inactivated. The choice of the inactivated copy is random. Briefly, X chromosomes contain an X-inactivation center (Xic), which contains the Xist gene. Xist is a long non-coding RNA that is mono-allelically upregulated from one X-chromosome at the onset of inactivation. Xist coats the X-chromosome from which it is expressed, and is necessary for its inactivation in cis. Before inactivation, pluripotency factors (Nanog, Sox2, Oct4) repress Xist expression to low levels on both chromosomes. As differentiation is initiated, their concentration decreases under the threshold necessary to inactivate both Xist alleles. It has been proposed that symmetry breaking between the two active X chromosomes might occur via a transient interaction between the Xic regions (Masui et al. 2011), allowing a monoallelic upregulation of Xist, involving pluripotency factors (Navarro and Avner 2010; Augui et al. 2011). Low and high expressions of Xist on both chromosomes are then consolidated by hetero/euchromatin deposition marks, respectively, and it becomes independent of pluripotency factors. A similar mechanism might apply to sister chromatids of autosomes. One possibility is that sister chromatid centromeres are loaded with equal amounts of specific factors. In response to extracellular cues, the expression of such factors would decrease, and symmetry could be broken between sister chromatid centromeres via their transient pairing (Fig. 3b2). One chromatid would then be randomly defined as the ‘template’ DNA, the other as the ‘nascent’ DNA. The asymmetric distribution of such specific factors could, as in the case of the asymmetric progression of the replication fork, result in secondary asymmetries between sister chromatids such as asymmetric methylation patterns, histone distribution, or histone modifications.
Secondary asymmetry between sister chromatids
Strand-biased DNA methylation has been observed in Arabidopsis thaliana centromeres (Luo and Preuss 2003) and hepatocellular carcinoma within the Apc gene (Jain et al. 2011). Recently, members of the 10-11 translocation proteins (Tet1, Tet2, and Tet3 in mouse) were shown to oxidize 5-methylcytosines (5mC) into other derivatives such as 5-(hydroxy)methylcytosines (5hmC), 5-formyl-cytosines, and 5-carboxy-cytosines (Fu and He 2012). 5hmC was reported to be enriched in mouse ES cells and in the cerebellum (Kriaucionis and Heintz 2009; Tahiliani et al. 2009). It is still unclear whether 5hmC is a degradation intermediate of 5mC or a bona fide epigenetic mark. 5hmC might inhibit the repressive effect of 5mC by preventing binding of methyl-binding proteins (Valinluck et al. 2004), or it might recruit specific partners involved in gene regulation. 5hmC is believed to be an intermediate of passive DNA demethylation by replication. The 5mC modification is usually maintained on the nascent DNA strand after replication by DNA methylation maintenance enzymes such as Dnmt1. Provided that 5hmC is not maintained after replication, this might generate an asymmetry between template and nascent DNA strands (Fig. 3c2). The presence and function of 5hmC in cells performing TDSS, in addition to a potential role for the de novo DNA methylating enzymes Dnmt 3a and 3b, remain to be explored. Interestingly, a MeDIP-Seq protocol was described where information on strand specificity of (hydroxy)methylation was determined (Ficz et al. 2011).
Over three decades ago, it was proposed that old and new nucleosomes segregate asymmetrically during DNA replication in a cell line (Weintraub 1976). This was later revealed to be the result of an experimental artifact (Russev and Hancock 1982). Ironically, recent studies in Drosophila melanogaster showed that during asymmetric division of male germline stem cells (GSCs), the ‘old’ pool of histone H3 remained associated to the stem cell, while the neosynthesized pool was enriched in the differentiating daughter cell (Tran et al. 2012). Interestingly, the histone variant H3.3 was distributed in a symmetric fashion in these cells. Strikingly, ectopic activation of the JAK-STAT signaling pathway forced symmetric division of GSCs, and subsequent symmetric distribution of ‘old’ and ‘new’ pools of H3. It is therefore possible that these different pools could carry information that can distinguish sister chromatids. In addition, it has recently been shown that sex chromosomes, but not autosomes, are non-randomly segregated during asymmetric divisions of D. melanogaster male GSCs (Yamashita, review this issue). The relationship between non-random sister chromatid segregation and histone segregation remains to be investigated in this case. In other studies, it has been suggested that the histone variant H2A.Z is differentially deposited on template and non-template DNA strands, within immortalized murine fibroblasts conditionally dividing asymmetrically (Huh and Sherley 2011). In that study, H2A.Z was either detected specifically on template DNA strands or on both template and non-template DNA strands, depending on the immunodetection protocol, suggesting that there is a molecular cloaking of H2A.Z on non-template DNA strands. In addition, pericentric heterochromatin has been shown to be a biophysically elastic component of the mitotic apparatus (Bouck and Bloom 2007). Different chromatin compositions between sister chromatids within these regions (Fig. 3c3, c4) might then mediate different interactions with chromatid recognition and segregation systems. Furthermore, it has recently been shown that nucleosomes can exhibit asymmetric histone modifications, such as permissive H3K4me3 and repressive H3K27me3. Such bivalent marks, coupled to non-random redistribution of histone pools during replication, might allow the expression of specific loci regulation on sister chromatids (Voigt et al. 2012).
Whatever the mechanism of symmetry breaking between sister chromatids might be, old and new centromeres would need to play a critical role in regulating the expression status of master regulatory genes (Fig. 3d), thereby allowing cell fate decisions to be executed during subsequent cell divisions.
Crosstalk between centromeres and master regulatory genes?
According to the silent sister hypothesis, epigenetic differences might exist between sister chromatids, within centromeres and stem/commitment genes. Asymmetric recognition of centromeres would then be the basis for non-random DNA segregation and this would in turn lead to asymmetric gene expression and differential cell fates between daughter cells (Lansdorp 2007). If true, when does the differential regulation of those ‘master’ genes occur? Before or after mitosis? Before or after asymmetric epigenetic labelling of centromeres? In other words, is there a crosstalk between epigenetic asymmetries at centromeres and master regulatory genes? If this is the case, which epigenetic mark appears asymmetric and acts as the initiating event? Asymmetric epigenetic modification of specific loci (centromeres or master gene bodies) between sister chromatids has not been explored in cells dividing with TDSS. Recently, a powerful strand-specific sequencing protocol was developed that can address some of these issues (Falconer et al. 2012). Briefly, ES cells are cultured in presence of BrdU for one cell cycle, resulting in hemi-labelled DNA. DNA from single cells is extracted, and high-throughput sequencing libraries are prepared. Before sequencing, libraries are exposed to UV light in the presence of Hoechst 33258, which sensitizes BrdU-labelled DNA to UV photolysis. Remaining unlabelled DNA strands are then sequenced from single cells, and sister chromatid exchanges are mapped with unprecedented resolution. Together with chromatin immunoprecipitation or bisulfite conversion of unmethylated cytosines to uracil, this method might allow the identification of epigenetic differences between sister chromatids that are differentially labelled with BrdU. However, a sequencing strategy might not be useful for centromeres, given their repetitive nature and the difficulty in mapping sequencing reads originating from repeats.
Recognition of specific chromatids by the mitotic machinery
Once sister chromatids are asymmetrically labelled, how does the mitotic apparatus segregate them selectively? Sister chromatid segregation by the mitotic spindle apparatus involves interactions between microtubules from spindle poles, kinetochore proteins, and centromeres. Achieving non-random sister chromatid segregation could require functionally different spindle poles to connect specifically, via kinetochores, with ‘old’ or ‘new’ centromeres. Interestingly, kinetochore proteins were reported to be distributed asymmetrically among progeny of a mother cell in S. cerevisiae (Thorpe et al. 2009). This cell gives rise to phenotypically distinct mother (m) and bud (b) cells, each of which in turn gives rise to two daughter cells: ‘mm’/’mb’ and ‘bm’/’bb’, respectively. A ‘mother’ lineage can then be defined: the ‘m’ cell, giving rise to the ‘mm’ cell, which in turn gives rise to the ‘mmm’ cell. Strikingly, the kinetochore proteins remained associated with the mother lineage, being mostly transmitted from the ‘m’ cell to the ‘mm’, and then the ‘mmm’ daughter cells. Such an asymmetric inheritance of kinetochore proteins might be related to asymmetric labelling and recognition of sister chromatids. Given its role in the attachment of microtubules to centromeres, Aurora B kinase would be an interesting candidate to regulate this process. Furthermore, microtubules can serially attach the two kinetochores, one before and the other one after chromosomes align during metaphase (Skibbens et al. 1995). Hence, sister chromatids asymmetrically loaded with kinetochores proteins could be recognized in a sequential fashion, ensuring their inheritance within the same daughter cell.
The existence of nonequivalent spindle poles has also been reported in many systems. Replication of centrioles is semi-conservative, generating ‘old’ and ‘new’ centrosomes with different structural properties (Uzbekov and Prigent 2007). In D. melanogaster, it has been shown that centrosomes can be inherited in a defined manner. During asymmetric divisions of male GSGs, the mother centrosome remains in the stem cell (Yamashita et al. 2007). In contrast, the mother centrosome is inherited by the committed daughter cell during neuroblast divisions (Conduit and Raff 2010; Januschke et al. 2011). In mGSCs and neuroblasts, centrosomes are important regulators of the balance between self-renewal and differentiation (Yamashita et al. 2003; Gonzalez 2008; Rusan and Rogers 2009).
It has been proposed that a crosstalk could exist between centromeres and centrosomes, via cargo proteins shuttling between kinetochores and centrosomes during TDSS (Tajbakhsh and Gonzalez 2009). Such a cargo protein could ensure that daughter cells will inherit matching types of chromatids and centrosomes (e.g. ‘old’ chromatids with mother or daughter centrosome). In yeast, the APC-related protein Kar9 is preferentially recruited by the old spindle pole (Maekawa et al. 2003). It binds to Myosin2 and moves along actin cables towards the bud (Liakopoulos et al. 2003). Asymmetric distribution of Kar9 is maintained by Dynein (Grava et al. 2006). These factors might be candidates for the selective capture of some sister chromatids by microtubules of a given spindle pole (Fig. 3e).
Conclusions and perspectives
Since the discovery of the double helix, and the mode of semi-conservative replication of DNA to transmit genetic material to daughter cells, it had been assumed that chromosomes containing old and new DNA strands are distributed randomly. There is now compelling evidence in prokaryotes and eukaryotes that DNA molecules of identical sequences can be treated differently. Beyond this notion, that old and new DNA can be recognized and distributed in a regulated manner is in itself truly remarkable. Recent evidence that this phenomenon is directly associated with differential cell fates of daughter cells points to a novel mode of gene regulation that needs to be considered among the more established mechanisms reported to date. Future studies will need to determine if this coupling is absolute, and how the in vivo niche regulates TDSS. Other considerations include DNA replication during S phase, chromatid choice selection during metaphase, and when differential cell fates are initiated. How global information on a chromatid is associated with local regulation of gene expression remains a mystery. It will not be surprising if, in addition to the putative mechanisms evoked above, new players will enter the scene to fill in these gaps in our knowledge. Whether TDSS of all or a few chromosomes is restricted to stem cells, or more widely used in different contexts, including cancers, also remains to be determined. The employment of rigorous standards linking single cell analysis, true daughter cell identities, and nucleotide analogues is necessary to quell some of the debate, and to focus discussions on mechanisms regulating chromatid identity. Finally, a shift in thinking from experimentally seeking ‘immortality’ of DNA strands to assessing asymmetry punctually in different developmental and physiological contexts is critical to introduce an unbiased view in the field of non-random DNA segregation.
Abbreviations
- 5hmC:
-
5(hydroxy)methylcytosine
- BrdU:
-
5-Bromo-2′-deoxyuridine
- CldU:
-
5-Chloro-2′-deoxyuridine
- CO-FISH:
-
Chromosome orientation fluorescence in situ hybridization
- EdU:
-
5-Ethynyl-2′-deoxyuridine
- IdU:
-
5-Iodo-2′-deoxyuridine
- mGSC:
-
Male germline stem cell
- PCNA:
-
Proliferating cell nuclear antigen
- TDSS:
-
Template DNA strand cosegregation
References
Arcangioli B (1998) A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J 17:4503–4510
Armakolas A, Klar A (2006) Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science 311:1146–1149
Augui S, Nora EP, Heard E (2011) Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet 12:429–442
Bailey SM, Goodwin EH, Meyne J, Cornforth MN (1996) CO-FISH reveals inversions associated with isochromosome formation. Mutagenesis 11:139–144
Bermejo R, Lai MS, Foiani M (2012) Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell 45:710–718
Biressi S, Rando TA (2010) Heterogeneity in the muscle satellite cell population. Semin Cell Dev Biol 21:845–854
Bischoff R, Holtzer H (1970) Inhibition of myoblast fusion after one round of DNA synthesis in 5-bromodeoxyuridine. J Cell Biol 44:134–150
Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E (2011) DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 8:16–29
Bosnakovski D, Xu Z, Li W, Thet S, Cleaver O, Perlingeiro RCR, Kyba M (2008) Prospective isolation of skeletal muscle stem cells with a Pax7 reporter. Stem Cells 26:3194–3204
Bouck DC, Bloom K (2007) Pericentric chromatin is an elastic component of the mitotic spindle. Curr Biol 17:741–748
Bylund GO, Burgers PMJ (2005) Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol 25:5445–5455
Cairns J (2006) Cancer and the immortal strand hypothesis. Genetics 174:1069–1072
Cairns J (1975) Mutation selection and the natural history of cancer. Nature 255:197–200
Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG (2009) The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138:1083–1095
Compe E, Egly JM (2012) TFIIH:when transcription met DNA repair. Nature reviews. Mol Cell Biol 13:343–354
Conboy MJ, Karasov AO, Rando TA (2007) High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol 5:102
Conduit PT, Raff JW (2010) Cnn dynamics drive centrosome size asymmetry to ensure daughter centriole retention in Drosophila neuroblasts. Curr Biol 20:2187–2192
Dalgaard JZ, Klar A (1999) Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 400:181–184
Escobar M, Nicolas P, Sangar F, Laurent-Chabalier S, Clair P, Joubert D, Jay P, Legraverend C (2011) Intestinal epithelial stem cells do not protect their genome by asymmetric chromosome segregation. Nat Commun 2:258
Falconer E, Chavez EA, Henderson A, Poon SSS, McKinney S, Brown L, Huntsman DG, Lansdorp PM (2010) Identification of sister chromatids by DNA template strand sequences. Nature 463:93–97
Falconer E, Hills M, Naumann U, Poon SSS, Chavez EA, Sanders AD, Zhao Y, Hirst M, Lansdorp PM (2012) DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat Methods 9:1107–1112
Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473:398–402
Fijalkowska IJ, Jonczyk P, Tkaczyk MM, Bialoskorska M, Schaaper RM (1998) Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. PNAS 95:10020–10025
Fu Y, He C (2012) Nucleic acid modifications with epigenetic significance. Curr Opin Chem Biol 16:516–524
Gayraud-Morel B, Chrétien F, Tajbakhsh S (2009) Skeletal muscle as a paradigm for regenerative biology and medicine. Regen Med 4:293–319
Gonzalez C (2008) Centrosome function during stem cell division: the devil is in the details. Curr Opin Cell Biol 20:694–698
Grava S, Schaerer F, Faty M, Philippsen P, Barral Y (2006) Asymmetric recruitment of dynein to spindle poles and microtubules promotes proper spindle orientation in yeast. Dev Cell 10:425–439
Harvey RP, Tajbakhsh S (2012) Biased DNA segregation and cardiac stem cell therapies. Circ Res 111:827–830
Huh YH, King J, Cohen J, Sherley JL (2011) SACK-expanded hair follicle stem cells display asymmetric nuclear Lgr5 expression with non-random sister chromatid segregation. Sci Rep 1:176
Huh YH, Sherley JL (2011) Molecular cloaking of H2A.Z on mortal DNA chromosomes during nonrandom segregation. Stem Cells 29:1620–1627
Irintchev A, Wernig A (1987) Muscle damage and repair in voluntarily running mice: strain and muscle differences. Cell Tissue Res 249:509–521
Jain S, Chang TT, Hamilton JP, Lin SY, Lin YJ, Evans AA, Selaru FM, Lin PW, Chen SH, Block TM, Hu CT, Song W, Meltzer SJ, Su YH (2011) Methylation of the CpG sites only on the sense strand of the APC gene is specific for hepatocellular carcinoma. PLoS One 6:e26799
Januschke J, Llamazares S, Reina J, Gonzalez C (2011) Drosophila neuroblasts retain the daughter centrosome. Nat Commun 2:243
Kajstura J, Bai Y, Cappetta D, Kim J, Arranto C, Amario DD, Matsuda A, Bardelli S, Ferreira-martins J, Hosoda T, Anversa P (2012) Tracking chromatid segregation to identify human cardiac stem cells that regenerate extensively the infarcted myocardium. Circ Res 7:894–906
Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K (2005) Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19:489–501
Karnani N, Taylor C, Malhotra A, Dutta A (2007) Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res 17:865–876
Karpowicz P, Morshead C, Kam A, Jervis E, Ramunas J, Ramuns J, Cheng V, Van der Kooy D (2005) Support for the immortal strand hypothesis: neural stem cells partition DNA asymmetrically in vitro. J Cell Biol 170:721–732
Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, Jackson TL, Morrison SJ (2007) Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 449:238–242
Klar A (1994) A model for specification of the left-right axis in vertebrates. Trends Genet 10:392–396
Klar A (1987) Differentiated parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature 326:466–470
Kriaucionis S, Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930
Kuang S, Rudnicki MA (2008) The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med 14:82–91
Kunkel TA (2004) DNA replication fidelity. J Biol Chem 279:16895–16898
Lansdorp PM (2007) Immortal strands? Give me a break. Cell 129:1244–1247
Lefaucheur JP, Sebille A (1995) Basic fibroblast growth factor promotes in vivo muscle regeneration in murine muscular dystrophy. Neurosci Lett 202:121–124
Lew DJ, Burke DJ, Dutta A (2008) The immortal strand hypothesis: how could it work? Cell 133:21–23
Liakopoulos D, Kusch J, Grava S, Vogel J, Barral Y (2003) Asymmetric loading of Kar9 onto spindle poles and microtubules ensures proper spindle alignment. Cell 112:561–74
Liu W, Wen Y, Bi P, Lai X, Liu XS, Liu X, Kuang S (2012) Hypoxia promotes satellite cell self-renewal and enhances the efficiency of myoblast transplantation. Development 139:2857–2865
Luo S, Preuss D (2003) Strand-biased DNA methylation associated with centromeric regions in Arabidopsis. PNAS 100:11133–11138
Maekawa H, Usui T, Knop M, Schiebel E (2003) Yeast Cdk1 translocates to the plus end of cytoplasmic microtubules to regulate bud cortex interactions. EMBO J 22:438–449
Masui O, Bonnet I, Le Baccon P, Brito I, Pollex T, Murphy N, Hupé P, Barillot E, Belmont AS, Heard E (2011) Live-cell chromosome dynamics and outcome of X chromosome pairing events during ES cell differentiation. Cell 145:447–458
Meselson M, Stahl FW (1958) The replication of DNA in Escherichia coli. PNAS 44:671–682
Mitchell KJ, Pannérec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA (2010) Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol 12:257–266
Mizuno K, Miyabe I, Schalbetter SA, Carr AM, Murray JM (2013) Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493:246–249
Moss FP, Leblond CP (1971) Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec 170:421–435
Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ (2005) Characterization of Dicer-deficient murine embryonic stem cells. PNAS 102:12135–12140
Méchali M, Yoshida K, Coulombe P, Pasero P (2013) Genetic and epigenetic determinants of DNA replication origins position and activation. Curr Opin Genet Dev 23:1–8
Nakano S, Stillman B, Horvitz HR (2011) Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell 147:1525–1536
Navarro P, Avner P (2010) An embryonic story: analysis of the gene regulative network controlling Xist expression in mouse embryonic stem cells. Bioessays 32:581–588
Nielsen O, Egel R (1989) Mapping the double-strand breaks at the mating-type locus in fission yeast by genomic sequencing. EMBO J 8:269–276
Ogino H, Satou W, Fujii M, Suzuki T, He Y, Michishita E, Ayusawa D (2002) The human MYOD1 transgene is suppressed by 5-bromodeoxyuridine in mouse myoblasts. J Biochem 132:953–959
Ono Y, Boldrin L, Knopp P, Morgan JE, Zammit PS (2010) Muscle satellite cells are a functionally heterogeneous population in both somite-derived and branchiomeric muscles. Dev Biol 337:29–41
Potten CS, Owen G, Booth D (2002) Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci 115:2381–2388
Prindle MJ, Fox EJ, Loeb LA (2010) The mutator phenotype in cancer: molecular mechanisms and targeting strategies. Curr Drug Targets 11:1296–1303
Probst AV, Okamoto I, Casanova M, El Marjou F, Le Baccon P, Almouzni G (2010) A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev Cell 19:625–638
Quyn AJ, Appleton PL, Carey FA, Steele RJC, Barker N, Clevers H, Ridgway RA, Sansom OJ, Näthke IS (2010) Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 2:175–181
Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL (2001) Replication dynamics of the yeast genome. Science 294:115–121
Rambhatla L, Ram-Mohan S, Cheng JJ, Sherley JL (2005) Immortal DNA strand cosegregation requires p53/IMPDH-dependent asymmetric self-renewal associated with adult stem cells. Cancer Res 65:3155–3161
Rando TA (2007) The immortal strand hypothesis: segregation and reconstruction. Cell 129:1239–1243
Reznik M (1969) Thymidine-3H uptake by satellite cells of regenerating skeletal muscle. J Cell Biol 40:568–571
Rocheteau P, Gayraud-Morel B, Siegl-Cachedenier I, Blasco M, Tajbakhsh S (2012) A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 148:112–125
Rothstein R, Michel B, Gangloff S (2000) Replication fork pausing and recombination or “gimme a break”. Genes Dev 14:1–10
Rusan NM, Rogers GC (2009) Centrosome function: sometimes less is more. Traffic 10:472–481
Russev G, Hancock R (1982) Assembly of new histones into nucleosomes and their distribution in replicating chromatin. PNAS 79:3143–3147
Sambasivan R, Gayraud-Morel B, Dumas G, Cimper C, Paisant S, Kelly RG, Kelly R, Tajbakhsh S (2009) Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev Cell 16:810–821
Schultz E, Jaryszak DL, Valliere CR (1985) Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 8:217–222
Shibahara K, Stillman B (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96:575–585
Shinin V, Gayraud-Morel B, Gomès D, Tajbakhsh S (2006) Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol 8:677–687
Skibbens RV, Rieder CL, Salmon ED (1995) Kinetochore motility after severing between sister centromeres using laser microsurgery: evidence that kinetochore directional instability and position is regulated by tension. J Cell Sci 2548:2537–2548
Sotiropoulou PA, Candi A, Blanpain C (2008) The majority of multipotent epidermal stem cells do not protect their genome by asymmetrical chromosome segregation. Stem Cells 26:2964–2973
Sotiropoulou PA, Candi A, Mascré G, De Clercq S, Youssef KK, Lapouge G, Dahl E, Semeraro C, Denecker G, Marine JC, Blanpain C (2010) Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol 12:572–582
Steinhauser ML, Bailey AP, Senyo SE, Guillermier C, Perlstein TS, Gould AP, Lee RT, Lechene CP (2012) Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 481:516–519
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Tajbakhsh S (2009) Skeletal muscle stem cells in developmental versus regenerative myogenesis. J Intern Med 266:372–389
Tajbakhsh S, Gonzalez C (2009) Biased segregation of DNA and centrosomes: moving together or drifting apart? Nature reviews. Mol Cell Biol 10:804–810
Tajbakhsh S, Rocheteau P, Le Roux I (2009) Asymmetric cell divisions and asymmetric cell fates. Ann Rev Cell Dev Biol 25:671–699
Tedesco FS, Cossu G (2012) Stem cell therapies for muscle disorders. Curr Opin Neurol 25:597–603
Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M (1998) AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J 17:667–676
Thorpe PH, Bruno J, Rothstein R (2009) Kinetochore asymmetry defines a single yeast lineage. PNAS 106:6673–6678
Tran V, Lim C, Xie J, Chen X (2012) Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338:679–682
Uzbekov R, Prigent C (2007) Clockwise or anticlockwise? Turning the centriole triplets in the right direction! FEBS Lett 581:1251–1254
Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC (2004) Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 32:4100–4108
Voigt P, LeRoy G, Drury WJ, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D (2012) Asymmetrically modified nucleosomes. Cell 151:181–193
Waghmare SK, Bansal R, Lee J, Zhang YV, McDermitt DJ, Tumbar T (2008) Quantitative proliferation dynamics and random chromosome segregation of hair follicle stem cells. EMBO J 27:1309–1320
Weintraub H (1976) Cooperative alignment of nu bodies during chromosome replication in the presence of cycloheximide. Cell 9:419–22
White MA, Eykelenboom JK, Lopez-Vernaza MA, Wilson E, Leach DRF (2008) Non-random segregation of sister chromosomes in Escherichia coli. Nature 455:1248–1250
White RB, Biérinx AS, Gnocchi VF, Zammit PS (2010) Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol 10:21
Yamashita YM, Jones DL, Fuller MT (2003) Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 301:1547–1550
Yamashita YM, Mahowald AP, Perlin JR, Fuller MT (2007) Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 315:518–521
Yennek S, Tajbakhsh S (2013) DNA asymmetry and cell fate regulation in stem cells. Seminars in Cell and Developmental Biology (in press)
Acknowledgments
We thank members of the lab for helpful comments and are grateful for support from the Institut Pasteur, Agence Nationale de la Recherche (Laboratoire d'Excellence Revive, Investissement d'Avenir; ANR-10-LABX-73) and (ANR-06-BLAN-0039), and the Association pour la Recherche sur le Cancer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Helder Maiato and Yves Barral.
Rights and permissions
About this article
Cite this article
Evano, B., Tajbakhsh, S. Sorting DNA with asymmetry: a new player in gene regulation?. Chromosome Res 21, 225–242 (2013). https://doi.org/10.1007/s10577-013-9359-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-013-9359-7