
Abstract
The study was designed to evaluate efficacy and superiority of capecitabine/bevacizumab + vinorelbine (CAP/BEV/VIN) compared to CAP/BEV alone. Main purpose was to introduce a taxane-/anthracycline-free first-line treatment in advanced breast cancer (ABC), in order to avoid long-term toxicities. In this open-label, superiority, phase 3 trial, patients with HER2-negative ABC were randomized 1:1 to receive either oral CAP at 1000 mg/m2 [twice daily, days 1–14, q3w] plus intravenous BEV at 15 mg/kg [day 1, q3w] (arm A) or in addition to this protocol intravenous VIN at 25 mg/m2 [days 1 + 8, q3w] (arm B) until disease progression, unacceptable toxicity or withdrawal of consent. Between 26 February 2009 and 26 October 2012, we randomised 600 patients (arm A N = 300; arm B N = 300) from 57 German outpatient-centres and 2 university hospitals. Median progression-free survival (PFS) (primary endpoint) was not improved with VIN (CAP/BEV, 8.8 months; CAP/BEV/VIN, 9.6 months; HR 0.84 [95 % CI 0.70–1.01], P = 0.058). Median overall survival (OS) (secondary endpoint) was 25.1 and 27.2 months for CAP/BEV and CAP/BEV/VIN, respectively, average HR 0.85 [95 % CI 0.70–1.03], P = 0.104). The 1- and 2-year OS rates appeared to be similar (78.0 and 77.0 %; 53.0 and 54.0 %). Toxicity profiles were generally mild and manageable. Adverse events occurred more frequently in arm B. Regarding the balance between clinical efficacy (PFS, OS) and toxicity, the CAP/BEV combination provides a favourable treatment option in first-line ABC avoiding taxane- and/or anthracycline-induced long-term toxicity. Superiority of CAP/BEV/VIN was not met, and side effects were even enhanced. Nevertheless, no safety issues occurred.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Key message
The CARIN trial is a large randomized phase 3 trial evaluating capecitabine/bevacizumab with or without vinorelbine in first-line treatment of ABC. PFS and OS are encouraging in both arms. Since the triple combination did not meet superiority, we suggest capecitabine/bevacizumab is favourable and recommendable, as OS is encouraging and taxane-/anthracycline-induced long-term toxicities can be avoided.
Introduction
Despite many new treatment options, advanced breast cancer (ABC) remains essentially incurable. Taxanes, especially paclitaxel (PAC), and anthracyclines represent standard agents in first-line chemotherapy [1, 2]. Unfortunately, taxanes and anthracyclines are associated with substantial side effects, including peripheral neuropathy, myelosuppression, cardiotoxicity, and hair loss [3, 4] that heavily impair patients’ performance and quality of life [5, 6]. Prolonged taxane and anthracycline exposure is not feasible because of cumulative toxic effects [7]. Therefore, capecitabine (CAP)-based combinations provide an effective and less toxic alternative for patients without rapidly progressive disease [8].
The addition of bevacizumab (BEV) to first-line chemotherapy in the E2100, AVADO, and RIBBON-1 trials resulted in prolongation of progression-free survival (PFS) and improved overall response rates (ORR) as compared to chemotherapy alone [9–11]. The placebo-controlled RIBBON-1 trial [11] was the first study demonstrating significantly improved ORR and PFS by adding BEV to first-line chemotherapy with either taxane-/anthracycline or CAP -based treatment. The TURANDOT trial [12, 13], a randomised phase 3 non-inferiority head-to-head study, assessed efficacy of BEV in combination with either PAC or CAP in first-line treatment for HER2-negative ABC.
Recently presented final OS data indicated non-inferiority of CAP/BEV (demonstrated in the stratified per-protocol analysis, supported by the stratified intent-to-treat analysis but not in the unstratified analysis). The OS curves seemed much the same, both in the per-protocol or the intent-to-treat analysis [13]. Although both, the TURANDOT and RIBBON-1 trial, demonstrated improved PFS for taxanes in combination with BEV, this did not translate into survival benefit.
Vinorelbine (VIN) monotherapy, evaluated in several clinical trials after failure of taxane-/anthracycline-based first-line metastatic treatment, yielded ORRs of about 29 % [14, 15]. VIN in combination with CAP revealed promising clinical activity and good tolerability in the neoadjuvant as well as in the metastatic setting [16–18]. Overlapping toxicities of both substances were rare.
CARIN was developed to improve efficacy through combination of VIN with CAP/BEV, thus offering an effective taxane-/anthracycline-free treatment option in first-line therapy of ABC. Primary objective was to demonstrate clinical superiority of CAP/BEV/VIN compared to CAP/BEV in terms of PFS. Secondary objectives included ORR, safety, and OS.
Methods
Patients
Eligible patients had HER2/-negative measurable or non-measurable disease, inoperable locally recurrent or ABC, no previous chemotherapy for advanced disease, and ECOG performance status ≤2; were aged ≥18 years; and had no sign of brain metastases. Adjuvant chemotherapy with either CAP or BEV or VIN was allowed if completed at least 12 months before randomisation. Further inclusion criteria comprised adequate liver, renal, cardiac, and haematological function; no uncontrolled hypertension; or proteinuria. All patients provided written informed consent. Independent ethics committees at all participating sites approved the protocol and all modifications.
Study design
CARIN was an open-label, randomised phase 3 trial. 600 patients were randomly assigned (1:1) to receive either CAP/BEV (arm A) or CAP/BEV/VIN (arm B). Randomisation was stratified by prior (neo)adjuvant therapy with taxanes or anthracyclines (yes/no) and hormone receptor status (±).
In both arms, CAP was administered orally at 1000 mg/m2 (twice daily, days 1–14, q3w), combined with intravenous BEV at 15 mg/kg (day 1, q3w). In arm B, intravenous VIN was added to CAP/BEV at 25 mg/m2 (days 1 + 8, q3w). Treatment was continued until progression of disease (PD), unacceptable toxicity, or withdrawal of consent. If any drug was discontinued for reasons of toxicity, treatment continued on the reduced regimen at allocated dosages. No BEV dose reduction was permitted, but treatment could be delayed. Beyond progression, all patients were offered standard-of-care treatment.
Efficacy and safety assessments
Tumour assessments according to RECIST 1.0 were performed at baseline, thereafter every 9 weeks until PD. After PD, patients were followed up for survival every 3 months over up to 3 years after last patient in.
Safety and tolerability assessments in terms of routine laboratory parameters, urinalysis, and vital signs were performed on a regular basis every cycle. Adverse events (AEs) were reported systematically throughout the study, including a 30-day safety follow-up period after treatment discontinuation. Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria, v3.0 and classified according to MedDRA v17.0 coding.
Statistical analysis
PFS as the primary objective was calculated from the date of randomisation to the date of first signs of tumour progression or death from any cause. Patients not experiencing PD or death were censored at the date of either last visit or start of new antineoplastic treatment. Calculating a drop-out rate of 10 %, a total of 600 patients (α = 0.05, two-sided, power = 80 %) were to be enrolled. At study data cut-off for analysis, less events than expected were observed, reducing the power to detect the initially calculated PFS difference between treatment arms (8.0 vs. 10.3; HR = 0.78) to 75 %. Secondary endpoints included ORR, OS, and safety.
Treatment effects on PFS were calculated and compared between treatment arms and within subgroups using the Kaplan–Meier and Cox regression method. Hazard ratios (HR) and 95 % confidence intervals (CI) were estimated by Cox proportional hazards analysis. Subgroup analyses were considered exploratory, no alpha adjustment for multiple testing was applied to the eight comparisons of subgroups: ‘age (<65 vs. ≥65 years)’, ‘number of metastatic sites (<3 vs. ≥3 sites)’, ‘prior taxane/anthracycline (yes/no)’, ‘visceral disease (involved/not involved)’, ‘triple negative breast cancer (TNBC yes/no)’, ‘ECOG performance status (0 vs. 1/2)’, ‘prior palliative endocrine therapy (yes/no)’, and ‘bone metastases (yes/no)’.
Since the OS curves for treatment comparison appeared non-proportional, average hazard ratios were determined by weighted Cox regression method [19]. For OS subgroup analyses, the Cox proportional hazards were estimated.
For objective response evaluation, treatment groups were compared using Cochran Mantel–Haenszel test. Patients were considered evaluable for response if they had measurable disease at baseline.
Demographic and clinical characteristics were evaluated descriptively. All statistical analyses were performed using Statistica v10.0 and R v3.2.0.
Results
Patients
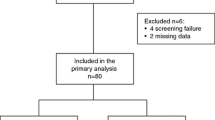
Between 26 February 2009 and 26 October 2012, 600 patients with locally advanced disease or ABC from 59 German outpatient centres and university hospitals were randomised, and 592 were eligible for efficacy and safety analysis (arm A, N = 297; arm B, N = 295). Eight patients did not receive the allocated treatment. The main reason for treatment discontinuation was PD (arm A, 179 [60.3 %]; arm B, 137 [46.4 %]). Treatment was permanently discontinued due to AE in 48 (16.1 %) and in 75 (25.4 %) patients in arms A and B, respectively (Fig. 1, [trial profile]).

Trial profile
Patient demographic and clinical baseline characteristics were generally well balanced between both arms (Table 1). Notably, the full analysis population was characterized by a considerable portion of patients older than 65 years (arm A: 105 [35.4 %]; arm B 132 [44.7 %]). The majority of patients was heavily pre-treated with (neo)adjuvant chemotherapy (arm A: 193 [65.0 %]; arm B: 195 [66.1 %]), including 114 (38.4 %) and 95 (32.2 %) patients with prior (neo)adjuvant taxane treatment in arm A and arm B, respectively. The proportion of patients with TNBC was identical in both arms (arm A: 61 [20.5 %]; arm B: 61 [20.7 %]).
Treatment exposure
Median duration of treatment was comparable between both arms (arm A: 27.9 weeks; arm B: 29.0 weeks). Median dose intensities for CAP were 84 and 79 % and for BEV 98 and 94 % in arms A and B, respectively. VIN dose intensity was 85 % (data not shown).
Efficacy
The addition of VIN slightly increased median PFS compared to CAP/BEV alone (8.8 vs. 9.6 months; HR 0.84 [95 % CI 0.70–1.01], log-rank P = 0.058), and therefore, the criteria for superiority of CAP/BEV/VIN were not met (Fig. 2, [patient characteristics]).

Kaplan–Meier estimate for progression-free survival (Intent-to-treat population). Arm A: capecitabine/bevacizumab; arm B: capecitabine/bevacizumab/vinorelbine
Hence, potential PFS differences between treatments were observed. When exploring subgroups according to clinical characteristics, significantly improved PFS among patients aged <65 years (8.2 vs. 10.2 months, HR 0.73 [95 % CI 0.58–0.93], P = 0.009), with <3 metastatic sites (8.8 vs. 10.7 months, HR 0.74 [95 % CI 0.59–0.93], P = 0.009), with (neo)adjuvant taxane/anthracycline pre-treatment (7.2 vs. 9.6 months, HR 0.71 [95 % CI 0.56–0.90], P = 0.004), with non-visceral disease (9.2 vs. 12.0 months, HR 0.63 [95 % CI 0.42–0.93], P = 0.020), with TNBC (4.2 vs. 7.0 months, HR 0.57 [95 % CI 0.39–0.84], P = 0.004), and with bone-only metastases (12.6 vs. 15.4 months, HR 0.46 [95 % CI 0.24–0.88], P = 0.017) was observed (Fig. 3, [subgroup analysis of progression-free survival]).

Subgroup analysis of progression-free survival according to baseline characteristics (intent-to-treat population). HR = hazard ratio; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group performance status; TNBC = triple-negative breast cancer. Arm A: capecitabine/bevacizumab; arm B: capecitabine/bevacizumab/vinorelbine
Confirmed ORRs were significantly lower in arm A compared to arm B (36.3 vs. 47.5 %, P = 0.047). Among responders, median duration of response was fairly comparable between arm A and arm B (8.8 vs. 9.6 months, HR 0.99 [95 % CI 0.67–1.45], P = 0.944) (data not shown).
At a median follow-up of 22.2 and 23.6 months in arm A and arm B, respectively, in total, 418 (70.6 %) patients had died (arm A: 218 [73.4 %]; arm B: 200 [67.8 %]) (Fig. 4). The 1- and 2-year OS rates were quite similar between both treatment arms (arm A: 77.6 and 53.3 %; arm B: 76.6 and 54.4 %) (data not shown). Also, median OS appeared comparable in arm A and arm B, respectively (25.1 vs. 27.2 months; Average HR 0.85 [95 % CI 0.70–1.03], P = 0.104). Nevertheless, treatment effects on OS became obvious comparing arm A versus arm B, respectively, within subgroups with taxane/anthracycline pre-treatment (21.5 vs. 25.2 months, HR 0.70 [95 % CI 0.54–0.90], P = 0.0048), and with non-visceral disease (29.3 vs. 42.4 months; HR 0.64 [95 % CI 0.51–0.98], P = 0.040) (Fig. 4, [Kaplan–Meier estimate for overall survival]).

Kaplan–Meier estimate for overall survival, separated by pre-defined subgroups (intent-to-treat population). a Overall population; b taxane and/or anthracycline pre-treated subgroup; c non-visceral disease subgroup. Arm A: capecitabine/bevacizumab; arm B: capecitabine/bevacizumab/vinorelbine
Safety
Common grade 3/4 events were (N patients [%], arm A vs. B) as follows: Nausea/vomiting (8 [2.7 %] vs. 19 [6.4 %]), Infection (27 [9.1 %] vs. 34 [11.5 %]), Fatigue/malaise (6 [2.0 %] vs. 19 [6.4 %]), Thromboembolic events (12 [4.0 %] vs. 15 [5.1 %]) including pulmonary embolism (4 [1.3 %] vs. 14 [4.8 %]), and peripheral neuropathia (1 [0.3 %] vs. 11 [3.7 %]) (Table 2b, [adverse events], separated by grades 3 and 4).
Conversely, grade 3/4 hand-foot syndrome was reported more often in arm A (70 [23.6 %] vs. 43 [14.6 %]), diarrhoea was reported slightly more often (13 [4.3 %] vs. 9 [3.0 %]), and hypertension occurred clearly more frequently in arm A (23 [7.8 %] vs. 9 [3.1 %]). Mucosal inflammation was reported equally (8 [2.7 %]) in both arms. Haematological toxicities as neutropenia (3 [1.0 %] vs. 57 [19.3 %]), leukopenia (1 [0.3 %] vs. 31 [10.5 %]), and febrile neutropenia (2 [0.7 %] vs. 5 [1.7 %]) were considerably more frequently reported in arm B. Overall, AEs of grade 3/4 (173 [58.2 %] vs. 216 [73.2 %]), serious AEs (112 [37.7 %] vs. 146 [49.5 %]), and AEs leading to treatment discontinuation occurred more often in arm B (Table 2a, [Overview of safety results]). In arm A, respectively, 63 (21.2 %) and 65 (21.9 %) patients required BEV and CAP treatment discontinuations due to AEs. In arm B, reflecting the higher incidence of AEs, more patients discontinued BEV (91 [30.8 %]), CAP (89 [30.2 %]), and VIN (100 [33.9 %]) treatment.
Serious AEs, deemed to be treatment-related by the investigator, led to death of three patients in arm A (pancytopenia, thromboembolic event, pulmonary embolism), and of two patients in arm B (pulmonary embolism and leukopenia associated with sepsis) (data not shown).
Discussion
The CARIN trial, a German phase 3 study, aimed to improve efficacy of CAP/BEV by adding VIN to establish a less toxic alternative to taxane-/anthracycline-based first-line treatment. PFS was the primary endpoint assuming a difference of 2.3 months in favour of the CAP/BEV/VIN combination. In the unselected population, superiority of the triple combination was not met. Exploring subgroups, the effect was more pronounced.
Compared to other phase 3 studies, the CARIN median PFS for CAP/BEV (8.8 months) was in the range of that observed in the CAP/BEV cohort of the TURANDOT (8.1 months) [12, 13] and the RIBBON-1 (9.2 months) [11] trials. RIBBON-1 [11] was the first study investigating efficacy and safety of BEV versus placebo combined with different standard chemotherapy backbones to be chosen by investigators before random assignment. This led to a pronounced imbalance between the treatment arms regarding taxane pre-treatment, with 40 % in the CAP/BEV arm and only 15 % in the taxane/anthracycline arm, thus hampering comparability of results. ORR and median PFS were higher in each BEV combination. This effect was most obvious in the CAP/BEV arm. The TURANDOT [12, 13] trial investigated in a randomised fashion whether OS with CAP plus BEV would be non-inferior to PAC plus BEV. Although response rates and PFS were significantly higher for PAC-BEV, results of the final analysis did not point to a survival benefit [13].
The debate regarding the use of taxanes and/or anthracyclines in first-line treatment of ABC on OS is still ongoing. The CARIN trial confirmed the efficacy of the taxane-/anthracycline-free CAP/BEV combination with a median OS of 25.1 months, which is quite comparable to 26.1 months reported from the CAP/BEV arm in the TURANDOT trial. Differences in OS between CARIN and TURANDOT may be explained by a significant discrepancy regarding pre-treatment with adjuvant or neoadjuvant taxanes in TURANDOT and CARIN (20 and 34 %, respectively), pointing to a more favourable prognosis for TURANDOT patients, when compared to the patient population in the CARIN trial. Notably, the 1-year OS rate in the CAP/BEV treatment arms was fairly comparable between CARIN (78 %), TURANDOT (81 %), and RIBBON-1 (81 %) trials, respectively. The taxane-containing PAC-BEV treatment in TURANDOT not only revealed OS of 30.2 months but also enhanced peripheral neuropathy. OS for CAP/BEV/VIN was 27.2 months.
On closer inspection, the CARIN OS estimates showed divergent curve characteristics beyond 33 months. This divergence may represent delayed clinical efficacy of arm B. The divergence may also be due to the differently responding subgroups. Particularly, subgroups with taxane/anthracycline pre-treatment and without visceral involvement obtained clinically meaningful survival benefits in the CAP/BEV/VIN approach, confirming VIN’s therapeutic activity. Nonetheless, these subgroup findings require further examination in larger patient cohorts.
Regarding response induction, taxane/anthracycline-based combinations with BEV seem to be more effective than CAP/BEV [8, 9]. Patients presenting with life-threatening metastatic organ involvement may thus benefit from taxane-/anthracycline-based first-line treatment. However, since almost all patients are at risk of developing taxane-induced neurotoxicity [21] that impairs patients’ daily life performance and overall quality of life, taxane treatment should be reserved for more advanced stages.
Undoubtedly, vinca alkaloids can also induce characteristic peripheral neurotoxicity [22, 23]. In this aspect, VIN added toxicity to the CAP/BEV combination. Treatment discontinuations due to toxicities occurred more frequently within CAP/BEV/VIN as compared to CAP/BEV alone, suggesting that VIN toxicities are in some way more severe or protracted. However, in the CARIN VIN-containing arm, only 10 of 295 patients developed grade 3 and 1 patient grade 4 polyneuropathy. In general, AEs > grade 3 were rarely seen. Common side-effects of VIN and main dose limiting toxicities were neutropenia, as observed in other studies [15, 24, 25]. VIN did not cause profound thrombocytopenia. Other toxicities were mild to moderate and generally well manageable.
There was an also manageable increase in adverse events due to hand–foot syndrome. The incidence (all grades) was higher in the CAP/BEV arm than in the CAP/BEV/VIN arm in spite of comparable dose intensities in both arms. In CARIN, hand–foot syndrome was mostly responsible for discontinuations of CAP. However, as compared to the BEV-PAC treatment arm in TURANDOT, the proportion of treatment discontinuations due to toxic effects in our CAP/BEV/VIN arm was somewhat lower (34 and 38 %, respectively).
The safety profile of BEV was consistent with known side effects [26, 27] and did not lead to a significant increase in toxicity. Severe side effects were rare even in patients with long-term treatment.
Conclusion
The CAP/BEV combination is an effective treatment option in first-line ABC, regarding PFS (8.8 months) and OS (25.1 months). The triple combination CAP/BEV/VIN did not meet superiority criteria and side effects were enhanced. The risk-/benefit-ratio were balanced in CAP/BEV as taxane/anthracycline-induced long-term toxicities can be avoided.
References
Zheng R, Han S, Duan C et al (2015) Role of taxane and anthracycline combination regimens in the management of advanced breast cancer: a meta-analysis of randomized trials. Med (Baltimore) 94:e803. doi:10.1097/MD.0000000000000803
Telli ML, Carlson RW (2009) First-line chemotherapy for metastatic breast cancer. Clin Breast Cancer 9(Suppl 2):S66–72. doi:10.3816/CBC.2009.s.007
Ghersi D, Willson ML, Chan MMK, et al. (2015) Taxane-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev 6: CD003366. doi: 10.1002/14651858.CD003366.pub3
Windebank AJ, Grisold W (2008) Chemotherapy-induced neuropathy. J Peripher Nerv Syst 13:27–46. doi:10.1111/j.1529-8027.2008.00156.x
Hall E, Cameron D, Waters R et al (2014) Comparison of patient reported quality of life and impact of treatment side effects experienced with a taxane-containing regimen and standard anthracycline based chemotherapy for early breast cancer: 6 year results from the UK TACT trial (CRUK/01/001). Eur J Cancer 50:2375–2389. doi:10.1016/j.ejca.2014.06.007
Shimozuma K, Ohashi Y, Takeuchi A et al (2012) Taxane-induced peripheral neuropathy and health-related quality of life in postoperative breast cancer patients undergoing adjuvant chemotherapy: n-sas bc 02, a randomized clinical trial. Support Care Cancer 20:3355–3364. doi:10.1007/s00520-012-1492-x
Hurtz H-J, Tesch H, Göhler T, et al. (2014) Long-term side effects of curative treatment for breast cancer—data from the MaTox project. Oncol Res Treat 37 (suppl.5):110
Miles D, Zielinski C, Martin M et al (2012) Combining capecitabine and bevacizumab in metastatic breast cancer: a comprehensive review. Eur J Cancer 48:482–491. doi:10.1016/j.ejca.2011.12.007
Gray R, Bhattacharya S, Bowden C et al (2009) Independent review of E2100: a phase III trial of bevacizumab Plus Paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol 27:4966–4972. doi:10.1200/JCO.2008.21.6630
Miles DW, Chan A, Dirix LY et al (2010) Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol 28:3239–3247. doi:10.1200/JCO.2008.21.6457
Robert NJ, Diéras V, Glaspy J et al (2011) RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol 29:1252–1260. doi:10.1200/JCO.2010.28.0982
Lang I, Brodowicz T, Ryvo L et al (2013) Bevacizumab plus paclitaxel versus bevacizumab plus capecitabine as first-line treatment for HER2-negative metastatic breast cancer: interim efficacy results of the randomised, open-label, non-inferiority, phase 3 TURANDOT trial. Lancet Oncol 14:125–133. doi:10.1016/S1470-2045(12)70566-1
Zielinski CC, Lang I, Inbar M, et al. (2015) TURANDOT: First-line bevacizumab plus capecitabine shows non-inferior overall survival versus bevacizumab plus paclitaxel for metastatic breast cancer| Abstract in: 40th Annual Meeting of the European Society for Medical Oncology ESMO @ ECC; abstract no. 1800 Sept 25–29
Zelek L, Barthier S, Riofrio M et al (2001) Weekly vinorelbine is an effective palliative regimen after failure with anthracyclines and taxanes in metastatic breast carcinoma. Cancer 92:2267–2272
Mansour M, Mourad C (2013) Phase II study of single agent oral vinorelbine as first-line treatment in patients with HER-2 negative metastatic breast cancer. Cancer Chemother Pharmacol 72:429–435. doi:10.1007/s00280-013-2216-8
von Minckwitz G, Kummel S, Vogel P et al (2008) Neoadjuvant vinorelbine-capecitabine versus docetaxel-doxorubicin-cyclophosphamide in early nonresponsive breast cancer: phase III randomized gepartrio trial. JNCI J Natl Cancer Inst 100:542–551. doi:10.1093/jnci/djn085
Estévez LG, Batista N, Sánchez-Rovira P et al (2008) A phase II study of capecitabine and vinorelbine in patients with metastatic breast cancer pretreated with anthracyclines and taxanes. Clin Breast Cancer 8:149–154. doi:10.3816/CBC.2008.n.015
Welt A, von Minckwitz G, Oberhoff C et al (2005) Phase I/II study of capecitabine and vinorelbine in pretreated patients with metastatic breast cancer. Ann Oncol 16:64–69. doi:10.1093/annonc/mdi024
Schemper M, Wakounig S, Heinze G (2009) The estimation of average hazard ratios by weighted Cox regression. Stat Med 28:2473–2489. doi:10.1002/sim.3623
Hess D, Köberle D, Thürlimann B et al (2007) Capecitabine and vinorelbine as first-line treatment in elderly patients (> or = 65 Years) with metastatic breast cancer. Oncology 73:228–237. doi:10.1159/000127414
De Iuliis F, Taglieri L, Salerno G et al (2015) Taxane induced neuropathy in patients affected by breast cancer: literature review. Crit Rev Oncol Hematol. doi:10.1016/j.critrevonc.2015.04.011
Moudi M, Go R, Yien CYS, Nazre M (2013) Vinca alkaloids. Int J Prev Med 4:1231–1235
Rossi A, Gridelli C, Gebbia V et al (2003) Single agent vinorelbine as first-line chemotherapy in elderly patients with advanced breast cancer. Anticancer Res 23:1657–1664
Seo HY, Lee HJ, Woo OH et al (2011) Phase II study of vinorelbine monotherapy in anthracycline and taxane pre-treated metastatic breast cancer. Invest New Drugs 29:360–365. doi:10.1007/s10637-009-9357-y
Lorusso V, Crucitta E, Silvestris N et al (2003) A phase I study of capecitabine in combination with vinorelbine in advanced breast cancer. Clin Breast Cancer 4:138–141
Jassem J, Carroll C, Ward SE et al (2009) The clinical efficacy of cytotoxic agents in locally advanced or metastatic breast cancer patients pretreated with an anthracycline and a taxane: a systematic review. Eur J Cancer 45:2749–2758. doi:10.1016/j.ejca.2009.05.035
Bernard-Marty C, Cardoso F, Piccart MJ (2004) Facts and controversies in systemic treatment of metastatic breast cancer. Oncologist 9:617–632
Acknowledgments
We thank all the patients and their families, caregivers, and investigators who participated in this study. Further, we thank Dr Andrea Kiemen who provided medical writing service on behalf of iOMEDICO. This manuscript was presented in part at scientific congresses as follows: Abstract and poster presentations at the American Society of Clinical Oncology annual meeting, Chicago, June 3–7, 2011 (abstract 1044), at the 36th Annual San Antonio Breast Cancer Symposium, December 10–14, 2013 (P3-13-01) and at the 39th European Society for Medical Oncology meeting, Madrid, Spain, September 26–30, 2014 (359PD).
Funding
The study was designed, conducted, and sponsored by iOMEDICO in cooperation with the AIO (Arbeitsgemeinschaft Internistischer Onkologen, Germany). The sponsor and the AIO had full responsibility for the study design, data collection, statistical analysis, the interpretation of results, and the decision to publish. The corresponding author had final responsibility for the preparation and finalization of this manuscript. The study was supported by Roche Pharma AG (Grenzach-Wyhlen, Germany).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
The authors certify that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (e.g. employment/affiliations, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Anja Welt has received honoraria/consultation fee from Amgen, Eisai, Interplan, Novartis, Roche, iOMEDICO, Pierre Fabre, grant/research support from Novartis. Norbert Marschner has received honoraria and reimbursement of travel costs from Roche, and CARIN research grant from Roche. Christian Lerchenmüller has conducted research sponsored by Roche. Thomas Decker has received honoraria for congress speeches and participating in advisory boards from Roche. Claus-Christoph Steffens has received honoraria for participating in advisory boards from Roche, Amgen, GSK, and Sanofi. Andreas Köhler has received honoraria and reimbursement of travel costs from Roche, and he is currently conducting research sponsored by this company. Reinhard Depenbusch has conducted research sponsored by Roche. Sabine Busies has declared no conflicts of interest. Susanna Hegewisch-Becker has received honoraria for participating in advisory boards from Roche, Merck, Amgen, and Lilly.
Ethical approval
The study was approved by the appropriate institutional and/or national ethics committee. All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all individual participants included in this study.
Additional information
On behalf of the Breast Cancer Working Group of AIO (Arbeitsgemeinschaft Internistische Onkologie).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Welt, A., Marschner, N., Lerchenmueller, C. et al. Capecitabine and bevacizumab with or without vinorelbine in first-line treatment of HER2/neu-negative metastatic or locally advanced breast cancer: final efficacy and safety data of the randomised, open-label superiority phase 3 CARIN trial. Breast Cancer Res Treat 156, 97–107 (2016). https://doi.org/10.1007/s10549-016-3727-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-016-3727-x