
Abstract
Aim
The objective of this research was to determine the effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease (patients aged 2 to 18 years at symptom onset) by systematic review.
Methods
A systematic search was conducted according to a protocol designed a priori of bibliographic databases and search engines. Studies selected according to pre-specified criteria were assessed for quality and risk of bias using standardised appraisal tools. Data were reported according to PRISMA conventions (Liberati et al. in PLoS Med 6:e1000100, 2009) and synthesised using GRADE (Guyatt et al. in J Clin Epidemiol 64:380–382, 2011).
Results
Of 2537 titles screened, 1 case series and 16 case reports met the inclusion criteria. No studies reported on the impact of enzyme replacement therapy on the survival of juvenile-onset patients. Low level evidence found that respiratory function may improve or be maintained in the early months of therapy. Improved muscle function in the first 6 to 12 months was also suggested, but results may be confounded by natural development. Patients with less severe baseline status and treated at a younger age showed more response than patients with more severe baseline status, treated as adults.
Conclusions
Interpretation of the findings was hindered by the lack of good quality evidence. The available data suggests that some JOPD patients may benefit in the short term from ERT through improved muscle strength and a reduced need for assisted ventilation. A focus by clinicians on improved and more consistent evidence collection, and use of study designs tailored to rare conditions, would provide more definitive results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pompe disease (PD) is a lysosomal storage disorder, caused by a deficiency of acid-alfa-glucosidase (GAA). Defective lysosomes rupture, spilling contents into the cell, leading to an accumulation of cellular glycogen. This neuromuscular disease is characterised by progressive muscle hypotonia, deterioration of respiratory and cardiac function.
PD is a rare and severe genetic disorder inherited in an autosomal recessive mode, with a wide phenotypic variation affecting age groups from infants to adults. Two classical forms of PD—infantile and late-onset—are recognised in the literature, although many cases do not fit the clinical picture for classical disease and are classified as atypical (Kishnani et al. 2006; Toscano and Schoser 2013) (Bembi et al. 2008). The estimated collective frequency is 1 in 40,000 people, with the infantile form making up one thirds of the cases (Bembi et al. 2008).
Patients older than 2 years diagnosed with Pompe disease tend to have a varied amount of GAA activity and considerable heterogeneity in clinical presentation. This late-onset form of PD can fall into the category of either juvenile or adult, depending on the age at onset of symptoms (Bembi et al. 2008; Pascual 2009). The age of symptom onset in juvenile-onset Pompe disease (JOPD) varies in the literature from 1 to 2 years old at the lower threshold and 16 to 18 years at the upper threshold (Bembi et al. 2008; Toscano and Schoser 2013). Categorisation may be confused by the patient’s age at diagnosis, which can occur years before (through genetic characterisation) or after symptom onset (if symptoms are not recognised in the early PD stages). Symptoms at onset vary but can include signs of muscle weakness or decreased respiratory capacity (Winkel et al. 2005). Juvenile and adult forms have been found to be associated with the milder c.-32-13T > G genotype, with variation in disease severity being attributed to different haplotypes (Kroos et al. 2007).
There is little published data for the juvenile-onset group, and its natural course is less well characterised. A separate analysis of this group is justified as patients are still in developmental stages, have a tendency to more severe disease and may have a different response to enzyme replacement therapy (ERT) to patients with adult-onset of the disease.
Published data on the natural history of late-onset PD (juvenile and adult patients) suggests that the earlier the age at which symptoms of PD occur, the worse the prognosis is likely to be (Winkel et al. 2005). However, within each age group, there is wide variation in the course of the disease (Pascual 2009).
Prior to ERT, there was no specific treatment for PD, with supportive care the only option available. ERT with alglucosidase alfa (Myozyme®) was included in the Australian Register of Therapeutic Goods (ARTG) in March 2008, and has been subsidised through the Life Saving Drugs Program (LSDP) since 2010 for infantile-onset Pompe disease. Treatment for JOPD has been subsidised through the LSDP since 2014. A review of this policy decision was undertaken as part of a broader review of the LSDP.
To inform this program review, a systematic literature review was conducted to evaluate the safety and effectiveness of treating JOPD with alglucosidase alfa. No systematic review had previously been published that addresses JOPD separately from adult-onset Pompe disease. This systematic review addresses that deficiency.
Methods
A protocol for the systematic review was designed a priori. The peer-reviewed literature was searched for studies assessing the safety and effectiveness of alglucosidase alfa in JOPD patients, using broad search criteria to maximise the number of articles identified. Searches for published literature were performed on 4th March 2016. The search included Orphanet (www.orphanet.net), PubMed, Embase.com, the Cochrane Library, CINAHL and the Web of Science (Web of Knowledge). A full list of databases searched and the terms used to search the PubMed database can be seen in Supplementary Data A.
A formal grey literature search was conducted with Google Scholar. Pertinent reviews and included studies were pearled and snowballed to ensure that all relevant articles were identified.
Study selection criteria
The study selection criteria were pre-specified using a PICOFootnote 1 format. JOPD patients were defined a priori as those for whom disease-onset occurred between 2 and 18 years of age. Criteria were applied independently by two researchers, and any differences were resolved by consensus (selection criteria are listed in Supplementary Data B).
Studies were assessed for eligibility using a staged approach, whereby case reports were included if sufficient higher level evidence was not identified. The level of evidence was determined by the NHMRC Evidence Hierarchy for Interventional evidence (Merlin et al. 2009).
Articles not written in English would have been included if they had represented a higher level of evidence than that available in English. Literature identified as opinion pieces, editorials, conference abstracts or papers without a clear study design or a description of method and results were not included.
Documentation of the literature search
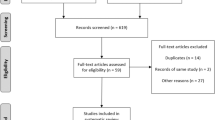
All literature identified in the search was documented using a flowchart recommended by the Preferred Reporting of Items in Systematic Reviews and Meta-analyses (PRISMA) statement (Liberati et al. 2009) (Fig. 1).

PRISMA flowchart for the literature identified on treatment for juvenile-onset Pompe disease (alglucosidase alfa)
Critical appraisal of selected evidence
It was intended that included studies would be critically appraised as per the protocol for risk of bias using appropriate appraisal tools for the different study designs (Khan et al. 2001; SIGN 2011), with the exception of case reports that are primarily descriptive in nature.
The body of evidence reporting on individual health outcomes was synthesised using the online Guideline Development Tool (McMaster University 2015) and rated according to the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) system (Guyatt et al. 2011).
Data extraction and synthesis
Data on the study authors, country and setting, population, intervention and comparator drug and dosage details, level of evidence, risk of bias, relevant outcome measures, results and follow-up period were extracted from the included studies. Characteristics of the study populations including age, race, sex, disease status and severity were reported. Key outcome data of the included studies were extracted in duplicate and independently by two researchers.
As there were insufficient data for a meta-analysis, the findings were synthesised into an overall narrative. This synthesis was informed by the GRADE method of evidence analysis (Guyatt et al. 2011). The findings were reported according to PRISMA reporting standards and incorporated all of the components required by AMSTAR for a high-quality systematic review (Shea et al. 2009).
Results
Systematic review findings
There was a paucity of comparative evidence identified in the literature specifically for juvenile Pompe patients. Studies that included a mixed late-onset population were only included if data from juvenile patients could be separated. One randomised controlled trial (Van Der Ploeg et al. 2010) and one systematic review (Toscano and Schoser 2013) were identified, which included mixed populations of late-onset PD, but as the data were combined, this ‘higher level’ evidence was not included. The remaining evidence was low level (case series and case reports) and lacking a relevant comparator.
One case series and 16 case report studies provided specific data on JOPD patients. Details of these studies are given in Supplementary Data C.Footnote 2
The case series (Bembi et al. 2010) included 24 late-onset PD patients, 7 of whom had JOPD. Outcomes were reported separately at baseline and 12-month intervals up to 3 years from the initiation of ERT for juvenile patients, and some data synthesis was performed. The case reports provided information on outcome measures before and after ERT for between one and five JOPD patients.
The age of onset for the JOPD patients varied from 2 to 17 years, with the age at first treatment with alglucosidase alfa varying from 2.3 to 52 years. One case report (Furusawa et al. 2014) did not state the dose of alglucosidase alfa, but all the other studies were consistent, using a dose of 20 mg/kg per fortnight.
Due to the high number of conference abstracts identified in the literature search, which then were not published in full as journal articles, the possibility of publication bias cannot be ruled out.
Effectiveness of ERT
Survival
There were no data specific to patients with JOPD, which addressed whether ERT would extend survival. Individual case reports mentioned age at death, but given the wide variability in the natural history of the disease, nothing could be concluded as to whether ERT extends life.
Quality of life
Three articles reported on the quality of life (QoL) for JOPD patients (Cerón-Rodríguez et al. 2014; Orlikowski et al. 2011; Winkel et al. 2004). Orlikowski et al. used the Short-form 36 (SF-36) to measure self-reported physical and mental health scores in two patients with advanced JOPD aged 16 and 14 years at symptom onset, and 28 and 40 years respectively when they started ERT. Both patients improved from baseline in the SF-36 mental component after 1 year of ERT. For the physical component, one patient’s score increased, while the other’s decreased (Supplementary Table S6). Changes were not large enough to be considered clinically meaningful.
In the other two studies (Cerón-Rodríguez et al. 2014; Winkel et al. 2004), there was no standardised measure for QoL; observations were reported narratively. All three patients in the studies showed signs of improved QoL, such as being able to attend school or longer periods sitting upright in a wheelchair, following one or more years of ERT.
Cardiorespiratory function
Cardiorespiratory function was reported in all but three of the included studies using a number of surrogate measures. The most commonly used were vital capacity (VC) and forced vital capacity (FVC).
VC was measured in seven case reports, two of which (Orlikowski et al. 2011; Papadimas et al. 2011) reported readings in the sitting and supine positions. Measurements taken at 4 to 6 months from baseline showed an increase in VC and FVC, with one exception in a patient whose VC remained unchanged while receiving ERT (Orlikowski et al. 2011). Both JOPD patients discussed by Orlikowski et al. (2011) had severe Pompe symptoms at baseline and remained on continuous ventilation support 24 h per day throughout the study.
At 1 year after the initiation of ERT, the results were more varied with some patients still measuring an increase in %VC, and others showing a decline. Measurements at 2 years or more after baseline were also inconsistent (Fig. 2a).

Graphical representation of the change in % predicted VC (A) and % predicted FVC (B) over time treated with ERT for individual JOPD cases. ERT, enzyme replacement therapy; FVC, forced vital capacity; JOPD, juvenile-onset Pompe disease; VC, vital capacity. Case notes A: case report 1 (Winkel et al. 2004) was reported as improved after 3 years of ERT from 14% (value not reported, reported here as 15%); measurements illustrated here for case reports 3 and 4 (Orlikowski et al. 2011) and 11 (Papadimas et al. 2011) were taken in the sitting position; case report 7 (Ishigaki et al. 2012) improved after 4 months ERT from 57 to 65% but was reported to return to baseline after 18 months (no value given, reported here as 57%); Korpel et al. (2009) case showed no improvement after 6 months ERT from 26% at baseline (value not reported). Case notes B: data for case reports 6 and 7 (Furusawa et al. 2012) were rounded to the nearest whole number for the purpose of reporting here
Winkel et al. (2004) performed a piecewise linear regression (“broken-stick” method) to compare the difference in the rate of change prior to receiving ERT, and after ERT. Patients 1 and 2 both had a significant decline in vital capacity in the 6 to 9 years prior to starting ERT (patient 1 r = −0.99, p < 0.001; patient 2, r = −0.98, p = 0.021). After the start of treatment, the slope of VC changed significantly (patient 1 p = 0.002; patient 2 p = 0.024), favouring the use of alglucosidase alfa.
FVC was measured in five patients from one study who were tested after 4 to 5 years of ERT (Deroma et al. 2014). FVC scores over 80% are considered to be within the normal range (Lachmann and Schoser 2013), so at baseline, three of the five patients were considered to have normal respiratory functioning. One patient who had much lower capacity at baseline than the others measured a significant increase in capacity over that time period (increase from 14 to 30%). The four other patients were within the normal range at follow-up (Fig. 2b).
Piecewise regression analysis was performed across case studies to assess the change in treatment effectiveness on %VC and FVC over the treatment period.Footnote 3 Time was initially partitioned into the intervals 0–6 months and > 6 months, and a separate line segment was fit to each interval for the outcome %VC and then FVC. Linear regression analysis showed no difference in treatment effect between the 0–6 month and > 6-month treatment periods for either measure when coefficients were compared (%VC − 0.02 vs. − 0.03, p = 0.996; FVC − 1.9 vs. 1.6, p = 0.624). Similarly, there was no difference between 0 and 12 month and > 12-month treatment periods. Due to low sample size (N = 22 for outcome %VC and N = 17 for outcome FVC), this piecewise regression analysis is likely to be underpowered. There was insufficient pre-ERT data to compare before and after treatment effects.
In some case reports, cardiorespiratory function was assessed through patient requirements for ventilation support. Ventilation hours were reduced moderately in one patient (aged 16 at treatment initiation) and remained consistent in another after 8 years of ERT (aged 32 at treatment initiation) (van Capelle et al. 2008). Changes to need for ventilation assistance showed improvement for three out of four patients after 12 months of ERT (age at treatment onset 17 to 44 years), as their status changed from ‘mild nocturnal dyspnoea’ to ‘normal’ (Kobayashi et al. 2010), but one patient deteriorated and required additional intermittent mechanical ventilation.
Gross motor function
Objective measures of muscle strength and function, such as the 6-min walk test or 10-m walk time (6MWT, 10MWT), global motor functioning and disability, gross motor functioning, muscle strength and motor testing were used in the case reports. The two most common assessments of gross-motor function were walk testing (6MWT and 10MWT tools) and muscle strength testing, using manual muscle testing, (MMT) and handheld dynamometer (HHD) tools.
Five articles published data on how far patients with JOPD could walk on the 6MWT and one article reported on the time taken for the 10MWT before and after treatment with alglucosidase alfa (Bembi et al. 2010; Deroma et al. 2014; Ishigaki et al. 2012; Korpel et al. 2009; Merk et al. 2009; van Capelle et al. 2010). Results for the 6MWT are summarised in Fig. 3.
Similar improvements were seen across most cases, with clinically important improvements observed over time,Footnote 4 particularly in the short term. The case series by Bembi et al. (2010) reported that seven juvenile patients (mean age at onset was 2.5 years) had an improvement in walking performance on the 6MWT over 3 years, with the greatest change seen in the first 12 months. After 3 years of alglucosidase alfa, JOPD patients were able to walk an average of 192 m further than at baseline. Similar improvements were reported by van Capelle (2010) (mean improvement 157.7 m) after 3 years and Deroma et al. (2014) (mean of 209.2 m of improvement) after 4–5 years of treatment. In all three studies, the patients were treated when they were aged between 5.9 and 15.2 years.
Two single case reports (Ishigaki et al. 2012; Korpel et al. 2009) found patients could walk further or longer after 4 and 6 months of ERT compared to baseline, but following that time, one case deteriorated and one case continued improving. There is little information on the natural history of JOPD, so it is difficult to know the extent to which improvements over time in a juvenile population are due to growth or to treatment benefit. There was no change in treatment effect over time when a piecewise regression was performed across the studies comparing the periods of 0–6 months and > 6 months of ERT treatment (6MWT coefficients − 42.9 vs. 2.4, p = 0.163) (Fig. 3).

Metres walked at baseline and follow-up after ERT using the 6MWT in case reports and a case series of patients with JOPD. 6MWT 6 minute walk test; ERT enzyme replacement therapy; JOPD juvenile onset Pompe disease. Notes: Results for the 6MWT in the case series reported by Bembi et al. (2010) is an average metres walked by 7 patients at base line and 12 months follow-up, and 6 patients for 2 and 3 year follow-up, the standard deviations were also reported (baseline 434.7 ± 260.8 m; 12 months 590.6 ± 213.6 m; 2 years 599.6±240.9; 3 years 614.3 ± 233.1 m; case report 7 (Merk 2008) indicated an inability to walk any distance at baseline or at 6 months follow-up; case report 8 (Ishigaki et al. 2012) 6MWT was measured at baseline and after 4 months ERT
Three studies used MMT to measure muscle strength. Despite considerable heterogeneity in baseline values between cases, all patients either remained stable, or improved while on ERT (Fig. 4a).

Change in muscle strength measured over time treated with ERT in individual JOPD cases measured by MMT (A) and HHD (B). ERT enzyme replacement therapy; HHD hand held dynamometer; JOPD juvenile onset Pompe disease; MMT manual Muscle Test. Notes A: MMT scored by an 11-point modified version of the Medical Research Council scale, % of maximum score (i.e. 0 to 100%); Notes B: van Capelle et al HHD values represent the sum score of the following muscle groups: neck flexion and extension, shoulder abduction, elbow flexion and extension, wrist extension, hip flexion and abduction, knee extension and flexion, ankle dorsiflexion and plantar flexion; Sugai et al HHD sumscore combines elbow extension and flexion, knee extension, key pinch and palmar pinch (Newtons)
One case report found muscle strength increased significantly to near normal values (van Capelle et al. 2010) in patients aged between 5.9 and 12.9 years at treatment. However, two other case reports (Furusawa et al. 2012; Ishigaki et al. 2012) did not find significant improvement in MMT after ERT. The two patients reported by Furusawa et al. had severe disease requiring ventilation, while a 10-year-old boy reported by Ishigaki et al. (2012) (first signs of JOPD at age 2) showed only minimal change on the MMT after 6 months of ERT compared to baseline.
Similar variable improvements in strength were seen in six patients when the HHD tool was used to measure muscle strength (Sugai et al. 2010; van Capelle et al. 2008, 2010) (Fig. 4b).
Of note, among the other measures used, is the quick motor function test (QMFT), a 16-item scale, which was developed specifically for use in patients with PD (van Capelle et al. 2010). The authors claim the QMFT provides an assessment of clinical severity and motor function and is also suitable for the longitudinal measure of changes over time in PD (van Capelle et al. 2012). All three patients described in van Capelle et al.’s 2010 article showed considerable improvement after 3 years of ERT using the QMFT (patients 1, 2 and 4 showed improvements of 70.3 to 95.3%, 73.4 to 92.2% and 67.2 to 92.2% of maximum possible, respectively).
Discussion
The JOPD population comprises a subset of the total PD population, that is likely to be larger but is under-diagnosed. Data on this subpopulation are rarely separated in the literature, despite the earlier onset being associated with disease prognosis (Winkel et al. 2005). The evidence we identified suggests that respiratory function may improve, or be maintained in the early months of ERT. After two or more years, the evidence is less consistent for respiratory function. Case reports also suggest that the need for assisted ventilation may be decreased with the use of ERT. Muscle functioning appears to improve with use of alglucosidase alfa, again with the largest improvements seen in the first 6 to 12 months of treatment. Improvements in gastrointestinal function and fatigue may also occur with the use of ERT (Bernstein et al. 2010; Sugai et al. 2010). The heterogeneity in the severity of symptoms between patients at baseline and the differences in the extent of treatment effect makes it difficult to determine which patients will do better than others. The limited nature of the evidence and the poor quality of the data prevents any generalisations or strong conclusions from being made regarding the effects of ERT and the characteristics that will predict patient response.
This systematic review highlights current challenges to the assessment of treatment for JOPD, some of which may be common to the translational of evidence science for rare diseases in general (Facey et al. 2014; Pariser and Gahl 2014). Recent research suggests ways in which measurement of the treatment effect of ERT for JOPD may be improved.
Previously, natural history survival data for patients with non-classic PD has been used to show that patients with later onset disease have significantly lower than normal life expectancy despite suffering a milder form than infantile-onset disease (Winkel et al. 2005). Natural history data have also proved to be a useful resource for assessing the effectiveness of treatment of JOPD. For example Kishnani et al. conducted a study of infantile PD patients through a retrospective chart review (Kishnani et al. 2006), which later provided a historical control for the assessment of ERT in that same population (Kishnani et al. 2009; Kishnani et al. 2007). Data collection by physicians and participation in rare disease registries and databases should be encouraged.Footnote 5
Accrued data has the potential to contribute to a greater understanding of the causes of clinical heterogeneity in JOPD. These differences between patients cannot be fully explained by mutation variability (Kroos et al. 2007) making it difficult to design eligibility criteria for controlled trials, especially with the small numbers of patients involved. To maximise the use of study data, it is valuable to describe in detail the context of treatment and symptoms in terms of time from diagnosis, time from onset of symptoms, disease severity, and age so that true changes can be measured between baseline and all time points.
Recently innovative trial designs for small populations have been discussed in the literature in regard to the assessment of many drugs for rare diseases reaching the market (Facey et al. 2014; Pariser and Gahl 2014). For JOPD, the challenge is to provide a valid estimate of treatment benefit and risk in a very small heterogeneous population without a traditional control population as a comparator. N-of-1 trials have a number of advantages for rare diseases, in particular in determining an individual treatment effect (Gabler et al. 2011). Milder forms of PD and older patients may have sufficiently stable JOPD to be assessed with an n-of-1 trial, but not those patients at the more severe end of the spectrum. Adaptive and sequential trials may be suitable for these JOPD patients, as all participants have the opportunity for active treatment, smaller sample numbers are required and trial times can be shortened if treatment effectiveness is established. The use of an historical control developed through a disease registry may still be the most effective means of establishing a treatment effect, as was used by Kishnani et al. (2007, 2009).
Patient reported outcomes (PROs) are becoming increasingly important in measuring the effectiveness of treatments for rare diseases. They can be designed to be more appropriate and sensitive to patient needs than traditional blunt measures, such as survival (Basch and Bennett 2014; Facey et al. 2014). The QMFT was developed by van Capelle for Pompe patients, to provide a sensitive measure of muscle function and disease progression that is quick and easy to use (van Capelle et al. 2012). The QMFT, which consists of 16 items based on clinical expertise, the Gross Motor Function Measure and the IPA/Erasmus MC Pompe survey was tested for scale in 91 patients and, while correlating well with other measures, it was sensitive to varying degrees of patient severity.
Conclusion
Poor quality evidence taken from case studies and one case series has suggested that ERT provides short-term benefits to some JOPD patients by improving muscle strength and function and reducing ventilation needs. Benefits may be greater when ERT is administered to those who are younger and those with less severe disease at baseline. Publication and reporting bias cannot be ruled out from influencing these findings, and the applicability of the results to the wider JOPD population cannot be assured.
The collection of data and assessment of ERT effectiveness in the JOPD population may be improved by increasing the use of global registries to document patient data in a systematic way and by using adaptive trial designs to randomise small patient numbers while enabling treatment for all. Further improvements in assessment of ERT may be gained by using sensitive PROs, specific to the disease.
Notes
Population; Intervention; Comparator; Outcomes
Analysis was performed using Stata Statistical Software: Release 14. College Station, TX: StataCorp LP.)
Minimum clinically important difference on the 6MWT is considered to be 54 m (Lachmann and Schoser 2013)
The Pompe Registry can be found at http://www.pompe.com/healthcare-professionals/pompe-registry.aspx
References
Basch E, Bennett A (2014) Patient-reported outcomes in clinical trials of rare diseases. J Gen Intern Med 29:S801–S803
Bembi B, Cerini E, Danesino C et al (2008) Management and treatment of glycogenosis type II. Neurology 71:S12–S36
Bembi B, Pisa FE, Confalonieri M et al (2010) Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J Inherit Metab Dis 33:727–735
Bernstein D, Bialer M, Mehta L, Desnick R (2010) Pompe disease: dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol Genet Metab 101:130–133
Cerón-Rodríguez M, Zamora A, Erdmenger J, Ureña R, Consuelo Sánchez A (2014) First case of a patient with late-onset Pompe disease: cardiomyopathy remission with enzyme replacement therapy. Boletín Médico del Hospital Infantil de México (English Edition) 71:41–46
Deroma L, Guerra M, Sechi A et al (2014) Enzyme replacement therapy in juvenile glycogenosis type II: a longitudinal study. Eur J Pediatr 173L:805–813
Facey K, Granados A, Guyatt G et al (2014) Generating health technology assessment evidence for rare diseases. Int J Technol Assess Health Care 30:416–422
Furusawa Y, Mori-Yoshimura M, Yamamoto T et al (2012) Effects of enzyme replacement therapy on five patients with advanced late-onset glycogen storage disease type II: a 2-year follow-up study. J Inherit Metab Dis 35:301–310
Furusawa Y, Mitsuhashi S, Mori-Yoshimura M et al (2014) Late-onset Pompe disease after 4 years of enzyme replacement therapy: an autopsy case. Neurol Clin Neurosci 2:7–9
Gabler N, Duan N, Vohra S, Kravitz R (2011) N-of-1 trials in the medical literature: a systematic review. Med Care 49:761–768
Guyatt G, Oxman A, Schunemann H, Tugwell P, Knottnerus A (2011) GRADE guidelines: a new series of articles in the Journal of Clinical Epidemiology. J Clin Epidemiol 64:380–382
Ishigaki K, Murakami T, Nakanishi T, Oda E, Sato T, Osawa M (2012) Close monitoring of initial enzyme replacement therapy in a patient with childhood-onset Pompe disease. Brain Dev 34:98–102
Khan K, Ter Riet G, Glanville J, Sowden A, Kleijnen J (2001) Undertaking systematic reviews of research on effectiveness. CRD's guidance for those carrying out or commissioning reviews. CRD Report Number 4 (second edition), NHS Centre for Reviews and Dissemination, University of York, York
Kishnani P, Hwu W, Mandel H, Nicolino M, Yong F, Corzo M (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148:671–676
Kishnani P, Corzo D, Nicolino M et al (2007) Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68:99–109
Kishnani P, Corzo D, Leslie N et al (2009) Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res 66:329–335
Kobayashi H, Shimada Y, Ikegami M et al (2010) Prognostic factors for the late onset Pompe disease with enzyme replacement therapy: from our experience of 4 cases including an autopsy case. Mol Genet Metab 100:14–19
Korpel M, Paeta A, Lofber M, Timonen M, Lamminen A, Kiuru-Enari S (2009) A novel mutation of the GAA gene in a Finnish late-onset Pompe disease patient: clinical phenotype and follow-up with enzyme replacement therapy. Muscle Nerve 40:143–148
Kroos M, Pomponio R, Hagemans M et al (2007) Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 68:110–115
Lachmann R, Schoser B (2013) The clinical relevance of outcomes used in late-onset Pompe disease: can we do better. Orphanet J Rare Dis 8:160
Liberati A, Altman D, Tetzlaff J et al (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med 6:e1000100
McMaster University (2015) GRADEpro GDT: GRADEpro Guideline Development Tool [Software]. (developed by Evidence Prime, Inc.). <https://gradepro.org/>
Merk T, Wibmer T, Schumann C, Kruger S (2009) Glycogen storage disease type II (Pompe disease)—influence of enzyme replacement therapy in adults. Eur J Neurol 16:274–277
Merlin T, Weston A, Tooher R (2009) Extending an evidence hierarchy to include topics other than treatment: revising the Australian ‘levels of evidence’. BMC Med Res Methodol 9:34–41
Orlikowski D, Pellegrini N, Prigent H et al (2011) Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord 21:477–482
Papadimas G, Spengos K, Konstantinopoulou A et al (2011) Adult Pompe disease: clinical manifestations and outcome of the first Greek patients receiving enzyme replacement therapy. Clin Neurol Neurosurg 113:303–307
Pariser A, Gahl W (2014) Important role of translational science in rare disease innovation, discovery, and drug development. J Gen Intern Med 29:S804–S807
Pascual S (2009) Phenotype variations in early onset Pompe disease: diagnosis and treatment results with Myozyme. Adv Exp Med Biol 652:39–46
Shea B, Hamel C, Wells G et al (2009) AMSTAR is a reliable and valid measurement tool to assess the methodological quality of systematic reviews. J Clin Epidemiol 62:1013–1020
SIGN (2011) SIGN 50: A guideline developer’s handbook. Scottish Intercollegiate Guideline Network, Edinburgh
Sugai F, Kokunai Y, Yamamoto Y et al (2010) Use of the muscle volume analyzer to evaluate enzyme replacement therapy in late-onset Pompe disease. J Neurol 257:461–463
Toscano A, Schoser B (2013) Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 260:951–959
van Capelle C, Winkel L, Hagemans M et al (2008) Eight years experience with enzyme replacement therapy in two children and one adult with Pompe disease. Neuromuscul Disord 18:447–452
van Capelle C, van der Beek N, Hagemans M et al (2010) Effect of enzyme therapy i juvenile patients with Pompe disease: a three-year open-label study. Neuromuscul Disord 20:775–782
van Capelle C, van der Beek N, de Vries J et al (2012) The quick motor function test: a new tool to rate clinical severity and motor function in Pompe patients. J Inherit Metab Dis 35:317–323
Van Der Ploeg A, Clemens P, Corzo D et al (2010) A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 362:1396–1406
Winkel L, Van den Hout J, Kamphoven J et al (2004) Enzyme replacement therapy in late-onset Pompe’s disease: a three-year follow-up. Ann Neurol 55:495–502
Winkel L, Hagemans M, van Doorn P et al (2005) The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 252:875–884
Funding
This study funded by the Department of Health, Australian Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
J. Milverton, S. Newton, and T. Merlin declare that they have no conflict of interest.
Research
As this was secondary research, no ethics approval or patients consent was required.
Additional information
Communicated by: Francois Feillet
Electronic supplementary material
ESM 1
(DOCX 64.6 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Milverton, J., Newton, S. & Merlin, T. The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: a systematic review. J Inherit Metab Dis (2018). https://doi.org/10.1007/s10545-018-0198-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10545-018-0198-8