
Abstract
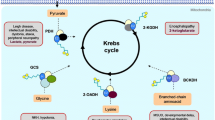
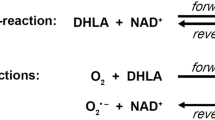
Lipoate is a covalently bound cofactor essential for five redox reactions in humans: in four 2-oxoacid dehydrogenases and the glycine cleavage system (GCS). Two enzymes are from the energy metabolism, α-ketoglutarate dehydrogenase and pyruvate dehydrogenase; and three are from the amino acid metabolism, branched-chain ketoacid dehydrogenase, 2-oxoadipate dehydrogenase, and the GCS. All these enzymes consist of multiple subunits and share a similar architecture. Lipoate synthesis in mitochondria involves mitochondrial fatty acid synthesis up to octanoyl-acyl-carrier protein; and three lipoate-specific steps, including octanoic acid transfer to glycine cleavage H protein by lipoyl(octanoyl) transferase 2 (putative) (LIPT2), lipoate synthesis by lipoic acid synthetase (LIAS), and lipoate transfer by lipoyltransferase 1 (LIPT1), which is necessary to lipoylate the E2 subunits of the 2-oxoacid dehydrogenases. The reduced form dihydrolipoate is reactivated by dihydrolipoyl dehydrogenase (DLD). Mutations in LIAS have been identified that result in a variant form of nonketotic hyperglycinemia with early-onset convulsions combined with a defect in mitochondrial energy metabolism with encephalopathy and cardiomyopathy. LIPT1 deficiency spares the GCS, and resulted in a combined 2-oxoacid dehydrogenase deficiency and early death in one patient and in a less severely affected individual with a Leigh-like phenotype. As LIAS is an iron–sulphur-cluster-dependent enzyme, a number of recently identified defects in mitochondrial iron–sulphur cluster synthesis, including NFU1, BOLA3, IBA57, GLRX5 presented with deficiency of LIAS and a LIAS-like phenotype. As in DLD deficiency, a broader clinical spectrum can be anticipated for lipoate synthesis defects depending on which of the affected enzymes is most rate limiting.



Similar content being viewed by others

References
Ajit Bolar N, Vanlander AV, Wilbrecht C et al (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet 22(13):2590–2602
Akiba S, Matsugo S, Packer L, Konishi T (1998) Assay of protein-bound lipoic acid in tissues by a new enzymatic method. Anal Biochem 258(2):299–304
Alfares A, Nunez LD, Al-Thihli K et al (2011) Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet 48(9):602–605
Autio KJ, Kastaniotis AJ, Pospiech H et al (2008) An ancient genetic link between vertebrate mitochondrial fatty acid synthesis and RNA processing. FASEB J 22(2):569–578
Baker PR, 2nd, Friederich MW, Swanson MA, et al (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137(Pt 2):366–379
Bowery NG, Smart TG (2006) GABA and glycine as neurotransmitters: a brief history. Br J Pharmacol 147(Suppl 1):S109–S119
Bunkoczi G, Pasta S, Joshi A et al (2007) Mechanism and substrate recognition of human holo ACP synthase. Chem Biol 14(11):1243–1253
Camaschella C, Campanella A, De Falco L et al (2007) The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110(4):1353–1358
Cameron JM, Janer A, Levandovskiy V et al (2011) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet 89(4):486–495
Campuzano V, Montermini L, Molto MD et al (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Carothers DJ, Raefsky-Estrin C, Pons G, Patel MS (1987) Rat liver mitochondria contain two immunologically distinct dihydrolipoamide dehydrogenases. Arch Biochem Biophys 256(2):597–605
Chen ZJ, Pudas R, Sharma S et al (2008) Structural enzymological studies of 2-enoyl thioester reductase of the human mitochondrial FAS II pathway: new insights into its substrate recognition properties. J Mol Biol 379(4):830–844
Chen Z, Kastaniotis AJ, Miinalainen IJ, Rajaram V, Wierenga RK, Hiltunen JK (2009) 17beta-hydroxysteroid dehydrogenase type 8 and carbonyl reductase type 4 assemble as a ketoacyl reductase of human mitochondrial FAS. FASEB J 23(11):3682–3691
Crooks DR, Jeong SY, Tong WH et al (2012) Tissue specificity of a human mitochondrial disease: differentiation-enhanced mis-splicing of the Fe-S scaffold gene ISCU renders patient cells more sensitive to oxidative stress in ISCU myopathy. J Biol Chem 287(48):40119–40130
Danhauser K, Sauer SW, Haack TB et al (2012) DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am J Hum Genet 91(6):1082–1087
Douglas P, Kriek M, Bryant P, Roach PL (2006) Lipoyl synthase inserts sulfur atoms into an octanoyl substrate in a stepwise manner. Angew Chem Int Ed Engl 45(31):5197–5199
Farhan SM, Wang J, Robinson JF et al (2014) Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol Genet Genomic Med 2(1):73–80
Feng D, Witkowski A, Smith S (2009) Down-regulation of mitochondrial acyl carrier protein in mammalian cells compromises protein lipoylation and respiratory complex I and results in cell death. J Biol Chem 284(17):11436–11445
Goodman SI, Frerman FE (1995) Organic acidemias due to defects in lysine oxidation:2-ketoadipic acidemia and glutaric acidemia. In: Valle D, Beaudet AL, Vogelstein B, et al. (eds) The online metabolic & molecular bases of inherited disease: McGraw-Hill, http://www.ommbid.com/. Accessed 25 Apr 2014
Haack TB, Rolinski B, Haberberger B et al (2013) Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis 36(1):55–62
Haviv R, Zeharia A, Belaiche C, Haimi Cohen Y, Saada A (2013) Elevated plasma citrulline: look for dihydrolipoamide dehydrogenase deficiency. Eur J Pediatr 45:265–269
Herbert AA, Guest JR (1968) Biochemical and genetic studies with lysine + methionine mutants of Escherichia coli: lipoic acid and alpha-ketoglutarate dehydrogenase-less mutants. J Gen Microbiol 53(3):363–381
Hermes FA, Cronan JE (2013) The role of the Saccharomyces cerevisiae lipoate protein ligase homologue, Lip3, in lipoic acid synthesis. Yeast 30(10):415–427
Hiltunen JK, Autio KJ, Schonauer MS, Kursu VA, Dieckmann CL, Kastaniotis AJ (2010) Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta 1797(6–7):1195–1202
Hiraga K, Kochi H, Hayasaka K, Kikuchi G, Nyhan WL (1981) Defective glycine cleavage system in nonketotic hyperglycinemia. Occurrence of a less active glycine decarboxylase and an abnormal aminomethyl carrier protein. J Clin Invest 68(2):525–534
Hoja U, Marthol S, Hofmann J et al (2004) HFA1 encoding an organelle-specific acetyl-CoA carboxylase controls mitochondrial fatty acid synthesis in Saccharomyces cerevisiae. J Biol Chem 279(21):21779–21786
Johnson MT, Yang HS, Magnuson T, Patel MS (1997) Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A 94(26):14512–14517
Kastaniotis AJ, Autio KJ, Sormunen RT, Hiltunen JK (2004) Htd2p/Yhr067p is a yeast 3-hydroxyacyl-ACP dehydratase essential for mitochondrial function and morphology. Mol Microbiol 53(5):1407–1421
Kollberg G, Tulinius M, Melberg A et al (2009) Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 132(Pt 8):2170–2179
Koyata H, Hiraga K (1991) The glycine cleavage system: structure of a cDNA encoding human H-protein, and partial characterization of its gene in patients with hyperglycinemias. Am J Hum Genet 48(2):351–361
Kure S, Kato K, Dinopoulos A et al (2006) Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat 27(4):343–352
Kursu VA, Pietikainen LP, Fontanesi F et al (2013) Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis in Saccharomyces cerevisiae. Mol Microbiol 90(4):824–840
Lim SC, Friemel M, Marum JE et al (2013) Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet 22(22):4460–4473
Mayr JA, Zimmermann FA, Fauth C et al (2011) Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am J Hum Genet 89(6):792–797
Mochel F, Knight MA, Tong WH et al (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet 82(3):652–660
Morikawa T, Yasuno R, Wada H (2001) Do mammalian cells synthesize lipoic acid? Identification of a mouse cDNA encoding a lipoic acid synthase located in mitochondria. FEBS Lett 498(1):16–21
Navarro-Sastre A, Tort F, Stehling O et al (2011) A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet 89(5):656–667
Odievre MH, Chretien D, Munnich A et al (2005) A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum Mutat 25(3):323–324
Olsson A, Lind L, Thornell LE, Holmberg M (2008) Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet 17(11):1666–1672
Packer L, Cadenas E (2011) Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr 48(1):26–32
Peng H, Shinka T, Inoue Y et al (1999) Asymptomatic alpha-ketoadipic aciduria detected during a pilot study of neonatal urine screening. Acta Paediatr 88(8):911–914
Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK (2013) Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol 48(1):67–72
Reed LJ (2001) A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276(42):38329–38336
Reed LJ, De BB, Gunsalus IC, Hornberger CS Jr (1951) Crystalline alpha-lipoic acid; a catalytic agent associated with pyruvate dehydrogenase. Science 114(2952):93–94
Schonauer MS, Kastaniotis AJ, Kursu VA, Hiltunen JK, Dieckmann CL (2009) Lipoic acid synthesis and attachment in yeast mitochondria. J Biol Chem 284(35):23234–23242
Shaag A, Saada A, Berger I et al (1999) Molecular basis of lipoamide dehydrogenase deficiency in Ashkenazi Jews. Am J Med Genet 82(2):177–182
Sloan JL, Johnston JJ, Manoli I et al (2011) Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet 43(9):883–886
Smith S, Witkowski A, Moghul A et al (2012) Compromised mitochondrial fatty acid synthesis in transgenic mice results in defective protein lipoylation and energy disequilibrium. PLoS One 7(10):e47196
Soreze Y, Boutron A, Habarou F et al (2013) Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J Rare Dis 8(1):192
Spiegel R, Saada A, Halvardson J et al (2013) Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur J Hum Genet 10:678
Stehling O, Lill R (2013) The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Med 3(7):1–17
Stuible HP, Meier S, Wagner C, Hannappel E, Schweizer E (1998) A novel phosphopantetheine:protein transferase activating yeast mitochondrial acyl carrier protein. J Biol Chem 273(35):22334–22339
Tort F, Ferrer-Cortes X, Thio M et al (2013) Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum Mol Genet 22:2590–2602
Van Hove J, Coughlin C, Scharer G (1993) Glycine Encephalopathy. In Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K eds. GeneReviews Seattle (WA).
Witkowski A, Thweatt J, Smith S (2011) Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J Biol Chem 286(39):33729–33736
Xu WY, Gu MM, Sun LH et al (2012) A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth disease type 2 in a large Chinese pedigree. Am J Hum Genet 91(6):1088–1094
Yi X, Maeda N (2005) Endogenous production of lipoic acid is essential for mouse development. Mol Cell Biol 25(18):8387–8392
Yoshino M, Koga Y, Yamashita F (1986) A decrease in glycine cleavage activity in the liver of a patient with dihydrolipoyl dehydrogenase deficiency. J Inherit Metab Dis 9(4):399–400
Zempleni J, Trusty TA, Mock DM (1997) Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr 127(9):1776–1781
Zhang L, Joshi AK, Smith S (2003) Cloning, expression, characterization, and interaction of two components of a human mitochondrial fatty acid synthase. Malonyltransferase and acyl carrier protein. J Biol Chem 278(41):40067–40074
Zhang L, Joshi AK, Hofmann J, Schweizer E, Smith S (2005) Cloning, expression, and characterization of the human mitochondrial beta-ketoacyl synthase. Complementation of the yeast CEM1 knock-out strain. J Biol Chem 280(13):12422–12429
Acknowledgments
Supported by the E-Rare project GENOMIT FWF I 920-B13, the Vereinigung zur Förderung Pädiatrischer Forschung und Fortbildung Salzburg, and the grant of the Fondo de Investigación Sanitaria (FIS) PI12/01138.
Conflict of interest
None.
Compliance with Ethics Guidelines
This article does not contain any studies on human subjects or on animals performed by any of the authors.
The first author designed the article; all authors contributed essentially to the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Garry Brown
Rights and permissions
About this article

Cite this article
Mayr, J.A., Feichtinger, R.G., Tort, F. et al. Lipoic acid biosynthesis defects. J Inherit Metab Dis 37, 553–563 (2014). https://doi.org/10.1007/s10545-014-9705-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-014-9705-8