
Abstract
Polyisoprenoid alcohols are membrane lipids that are present in every cell, conserved from archaea to higher eukaryotes. The most common form, alpha-saturated polyprenol or dolichol is present in all tissues and most organelle membranes of eukaryotic cells. Dolichol has a well defined role as a lipid carrier for the glycan precursor in the early stages of N-linked protein glycosylation, which is assembled in the endoplasmic reticulum of all eukaryotic cells. Other glycosylation processes including C- and O-mannosylation, GPI-anchor biosynthesis and O-glucosylation also depend on dolichol biosynthesis via the availability of dolichol-P-mannose and dolichol-P-glucose in the ER. The ubiquity of dolichol in cellular compartments that are not involved in glycosylation raises the possibility of additional functions independent of these protein post-translational modifications. The molecular basis of several steps involved in the synthesis and the recycling of dolichol and its derivatives is still unknown, which hampers further research into this direction. In this review, we summarize the current knowledge on structural and functional aspects of dolichol metabolites. We will describe the metabolic disorders with a defect in known steps of dolichol biosynthesis and recycling in human and discuss their pathogenic mechanisms. Exploration of the developmental, cellular and biochemical defects associated with these disorders will provide a better understanding of the functions of this lipid class in human.
Similar content being viewed by others
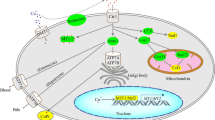


Structure of dolichol metabolites and their tissue and subcellular distribution
Polyisoprenoid alcohols are membrane lipids present in every cell, from archaea to higher eukaryotes. They all have in common a function as sugar carrier for protein glycosylation but display a surprising structural diversity between different species and also inside the same organism (Swiezewska and Danikiewicz 2005; Jones et al. 2009). The most common species, dolichol was originally identified by Pennock, Hemming and Morton in 1960 (Pennock et al. 1960). Dolichol phosphate requirement for glycoprotein biosynthesis was established 10 years later by Behrens and Leloir (Behrens and Leloir 1970). During dolichol biosynthesis, isoprenoids are used as a five-carbon building block to generate linear polymers. These polyisoprenoids are not present as species of a single chain length, but are found as a mixture (or family) of four or more different chain lengths, with one or two chain lengths predominating. The size of dolichols (i.e., the mixture of different chain lengths) is variable depending on the species. In the yeast S.cerevisiae, the size ranges from 14 repeating isoprene units to 18 (Dol-14 to Dol-18) and the major dolichols are Dol-16 and Dol-17. A preponderance of Dol-19 has been observed for several vertebrates including human (Chojnacki and Dallner 1988).
Whereas the length of these lipids is variable, the configuration of the double bonds of polyisoprenoids is fixed. All dolichol species characterized so far show a di-trans, poly-cis configuration. This is a consequence of the involvement of two types of enzyme, a short-chain trans-prenyl transferase and a long-chain cis-prenyl transferase (Skorupinska-Tudek et al. 2008). The α-terminal isoprene unit is unsaturated in polyprenol (formerly dehydrodolichol), whereas dolichol contains a saturated α-terminal unit (See Fig. 2). This saturation event is of great interest because it is absent in bacteria but required in many eukaryotic cells (Krag 1998). The terminal hydroxyl group of dolichol may exist either free, phosphorylated or esterified with fatty acids. Depending on the tissue or membrane, dolichyl esters represent 10 to 60% of the total dolichol content and Dol-P in general represents less than 10% (Tollbom and Dallner 1986).
Dolichol biosynthesis is known to occur mainly on the cytoplasmic face of the ER, where N-glycosylation occurs. Besides the presence of glycosylated dolichol species in the ER, dolichol is present in all subcellular membrane systems. The free alcohol, but also phosphorylated and esterified dolichol are detected in peroxisomes and are highly enriched in lysosomes, plasma membrane and Golgi vesicles (Rip et al. 1981). The ER microsomal fraction is low in dolichol compared to other organelles. The mitochondrial membranes and nuclei also contain a limited amount of dolichol, especially low in the mitochondrial inner membrane.
All tissues and almost all membranes in eukaryotic cells contain dolichol. In general, they are present in small absolute quantities, only a few percent compared to glycerol-based phospholipids (Chojnacki and Dallner 1988). However, the amount is highly variable depending on tissue and age (Rip et al. 1985). For example, in human endocrine tissues, this lipid is present in milligram quantities per gram of wet weight, as much as phospholipids. In different species, several organs are highly enriched in dolichol. In human, testes and especially pituitary are associated with high contents of dolichol. Surprisingly, human tissues seem to have levels 5 to 10-fold higher in comparison to the corresponding tissues from rat or mouse, ranging from ~ 0.1-2 mg/g fresh weight in human versus ~ 10-200 μg/g in mouse or rat (Rip and Carroll 1985). Dolichol is also detected in the serum at low concentration, associated with the HDL fraction (Elmberger et al. 1988).
The reason of the specific enrichment of some tissues versus others is unknown, but does not seem to be correlated with the level of glycosylation.
A significant age-related accumulation of dolichol has been reported. In the human brain, a 100-fold increase has been detected during the aging process (Pallottini et al. 2003), a similar increase has been observed in other tissues. These accumulations might also be involved in pathology as elevated dolichol levels have been detected in hepatocarcinogenesis and ceroid lipofuscinosis (Ericsson et al. 1993). However, this accumulation seems to be disrupted in Alzheimer’s disease as a significant reduction of dolichol levels has been detected in patient brains (Edlund et al. 1994).
Cellular functions of dolichol
Phosphorylated dolichol has a well defined role as a lipid carrier for the glycan precursor in the early stages of N-linked protein glycosylation. This precursor is assembled in the endoplasmic reticulum of all eukaryotic cells (Fig. 1). During N-linked protein glycosylation, a first dolichol phosphate is used by the GlcNAc-1-phosphotransferase to start the assembly of the lipid-linked oligosaccharide (LLO) on the cytoplasmic face of the ER. The product of this reaction is elongated to Man5GlcNAc2-P-P-Dol and then flipped to the inside of the ER. Then four other dolichol phosphates, previously linked to a mannose (Man-P-Dol) and flipped inside the ER lumen, are used as sugar donor for LLO synthesis. Finally, three dolichol phosphates in the form of dolichol phosphate glucose (Dol-P-Glc) as sugar donor are used for completion of the LLO before transfer of the glycan onto nascent proteins (Fig. 1). In addition to its role during LLO biosynthesis, Man-P-Dol is a necessary substrate for C- and O-mannosylation as well as GPI anchor biosynthesis (Rosenwald et al. 1990).

Dolichol cycle in the endoplasmic reticulum in human. The Dol-P pool available for glycosylation reactions originates from de novo synthesis of dolichol phosphate and its recycling from the lumenal leaflet to the cytoplasmic leaflet of the ER. Cellular functions of dolichol are indicated including known functions in N-glycosylation (1–2), O-mannosylation (3), GPI-anchor biosynthesis (4) and suspected effects on membrane biophysical properties (5–6). Abbreviations: FPP, farnesyl pyrophosphate; DHDDS, dehydrodolichyl diphosphate synthase; SRD5A3, steroid 5-alpha reductase 3; DOLK, dolichol kinase; DPM1-3, dolichyl-phosphate mannosyltransferase polypeptide 1–3; MDPU1, mannose-P-dolichol utilization defect 1; ALG5, asparagine-linked glycosylation 5; DOLPP1, dolichyl pyrophosphate phosphatase 1; LLO, lipid-linked oligosaccharide
In comparison to dolichol phosphate, the role of the free alcohol and its carboxylic ester is less established. An important question is the effect of these lipids on the physical properties of biological membranes. Biophysical studies have shown that dolichol and dolichol phosphates destabilize the bilayer structure of model membranes, and also increase their fluidity (Valtersson et al. 1985). Isolated membrane phospholipids may adopt different phases including the lamellar bilayer phase and a phase where phospholipids are arranged as cylinders with the lipid head group facing the inner core of the cylinder, also called hexagonal phase II (Turned and Gruner 1992). Dolichol promotes this hexagonal phase II organization, which may facilitate movements between the two layers of the membrane and may elicit the transmembrane movement of LLOs. This property would add another function for this lipid in the glycosylation process. Another interesting property is the fact that dolichol enhances vesicle fusion and that dolichol phosphate stimulates the fusion of rat liver microsomes (van Duijn et al. 1986). In this way, dolichol might play a more general role in membrane trafficking. In vivo, consequences of dolichol synthesis defects have been investigated in a yeast mutant for the cis-isoprenyltransferase (cis-IPTases) RER2 responsible for the elongation of the polyisoprenoid chain (Sato et al. 1999) (Belgareh-Touze et al. 2003). These studies suggest that free dolichol is involved in vesicle trafficking, a function in agreement with an enrichment of the Golgi apparatus for this lipid.
A specific role for the dolichol-ester has not been clearly established. Besides an effect on membrane properties, the esterified form was suggested to be necessary for intracellular dolichol transport (Tollbom and Dallner 1986; Turunen and Schedin-Weiss 2007). An oxidized derivative of dolichol, dolichoic acid has also been identified in human brain (Ward et al. 2007). It is speculated to result from dolichol catabolism (Van Houte et al. 1997), but its exact function is unknown.
De novo synthesis of dolichol and recycling
The first steps in the biosynthesis of dolichol are common with those for cholesterol and belong to the mevalonate pathway (Fig. 2). This pathway starts with the condensation of acetyl-CoA, followed by the production of mevalonate through the action of HMG-CoA reductase (Swiezewska and Danikiewicz 2005). After several modifications, isopentenyl pyrophosphate (IPP) is produced. IPP condensation gives rise to farnesyl pyrophosphate (FPP), used as the substrate of many reactions including the synthesis of cholesterol, dolichol, solanesyl pyrophosphate (i.e., the side chain of ubiquinone) and for isoprenylation of proteins.

De novo biosynthesis of dolichol phosphate in human. Polyisoprenoids are generated by the condensation of isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), the 5-carbon building blocks of all isoprenoids. The mevalonate pathway is responsible for the synthesis of IPP and DMAPP. The first step dedicated to dolichol biosynthesis is the elongation of farnesyl pyrophosphate by the dehydrodolichyl diphosphate synthase (DHDDS) . This cis-prenyl transferase uses IPP as substrates to produce polyprenol pyrophosphate. It is believe that this polyprenol pyrophosphate is then dephosphorylated into polyprenol before its reduction by SRD5A5. Finally, the dolichol kinase (DOLK) transfers a phosphate from CTP to dolichol
Elongation step
Cis-IPTases catalyse the synthesis of polyprenol pyrophosphate by sequential head-to-tail cis addition of multiple molecules of IPP to farnesyl pyrophosphate (FPP). This is the first step dedicated to dolichol synthesis and takes place on the cytoplamic side of the ER. At least two enzymes are involved in this process in yeast (i.e., RER2 and SRT1). In mammals only one cis-IPTase has been characterized (Shridas et al. 2003). However, some other uncharacterized or partially characterized enzymes might be involved. For example, the human protein NUS1, homolog of the yeast nuclear undecaprenyl pyrophosphate synthase 1, shares significant sequence homologies with cis-IPTs. Structural analysis of these enzymes (Takahashi and Koyama 2006) as well as functional studies in yeast (Sato et al. 1999) indicate that cis-IPTs themselves determine the chain length of their products. This was illustrated in a recent study from Rush et al. (Rush et al. 2010) where the yeast cis-IPTase was replaced by a bacterial one, resulting in the synthesis of a dolichol with the length of the bacterial polyisoprenoid (i.e., 11 units instead of 14–18 units). An intriguing aspect of these cis-IPTases is the absence of a predicted transmembrane domain in their sequence despite their ER localization. This might be explained by the existence of unknown membrane protein interactors.
Dephosphorylation step
This step is supposed to occur between the elongation process and the reduction step. Polyprenol pyrophosphate phosphatase and polyprenol phosphatase activities have been reported in whole-cell homogenates (Wolf et al. 1991), however, the molecular basis supporting these enzymatic reactions is unknown. A dolichol pyrophosphate phosphatase has been identified in yeast (CWH8) (Fernandez et al. 2001). This enzyme is involved in the recycling of dolichol after oligosaccharide transfer to a protein, by removing one phosphate from Dol-PP in the ER (Fig. 1). This recycling of dolichol phosphate plays a major role in the regulation of the dolichol phosphate pool available for the different glycosylation pathways (Schenk et al. 2001).
An alternative hypothesis suggests that the terminal event of the elongation process could be the condensation of a molecule of isopentenol instead of IPP (Ekstrom et al. 1987), avoiding a final dephosphorylation and reduction event.
Reduction step
Several mutant cell lines defective for this step have been identified, including Chinese hamster ovary cell (CHO) mutants belonging to the same complementation group lec9. They were identified based on lectin resistance and present a general hypoglycosylation of proteins associated with a reduced amount of mature LLO (Rosenwald and Krag 1990). These mutants show a similar phenotype with an accumulation of polyprenols of a chain length identical to dolichols usually present in these cells, but lacking the saturation of the alpha-isoprene unit. Polyprenols are obligatory intermediates for the synthesis of dolichols and are detected in large amount during in vitro biosynthesis of dolichol. However, in vivo, polyprenol levels are usually very low, often undetectable (Ekstrom et al. 1987; Sagami et al. 1993), probably the result of a fast conversion to dolichols. A mutant cell line from Trypanosoma Brucei shows similar biochemical defects including the hypoglycosylation of proteins and the accumulation of polyprenol (Acosta-Serrano et al. 2004). These CHO and Trypanosoma mutants have in common a defect only partial in dolichol biosynthesis, still present in these cells, and an unknown genetic basis for this defect.
We reported recently the identification of the first enzyme associated with this reduction step. This enzyme, SRD5A3 in human and DFG10 in S.cerevisiae, shows homology with steroid 5-alpha reductase enzymes (Cantagrel et al. 2010). Mutation in the corresponding genes results in a partial block of dolichol biosynthesis with an accumulation of polyprenol and hypoglycosylation of proteins. Human SRD5A3 can correct the phenotype of the yeast deleted for DFG10 by preventing the accumulation of polyprenols and the hypoglycosylation of carboxypeptidase Y (CPY). Mutations associated with this defect in human are mostly homozygous early truncating mutations, resulting in some cases in non-sense mediated mRNA decay. These observations suggest that null alleles are responsible for this condition. In yeast, the dfg10 deletion mutant is also only partially blocked in this reduction step with an important decrease in dolichol (i.e., a 70% decrease was detected (Cantagrel et al. 2010)) associated with polyprenol accumulation. These results reveal an unexpected alternative pathway for dolichol synthesis, probably conserved from yeast to human. The identification of these SRD5A3/DFG10 genes may be a starting point to look for physical and genetic interactors in order to understand better the coupling of this reduction step with polyprenol pyrophosphate elongation and dephosphorylation as well as the nature of the alternative pathway involved in the polyprenol reduction step.
Phosphorylation step
The availability of dolichol phosphate is one of the rate-limiting factors controlling the synthesis of LLOs in eukaryotic cells (Carson et al. 1981; Spiro and Spiro 1986). Dolichol Kinase (DOLK) is a CTP-dependent enzyme localized in the ER with a catalytic site oriented to the cytoplasmic side of the ER (Shridas and Waechter 2006). The yeast gene encoding for the dolichol kinase is essential, the phenotype resulting from a mutation of this gene was studied with the temperature sensitive mutant sec59 (Heller et al. 1992). In this mutant, Dol-P levels are less than 10% of wt yeast at restrictive temperature. This reduced availability of Dol-P results in incomplete glycosylation of secretory proteins and strongly affects O-mannosylation and GPI anchor synthesis (Heller et al. 1992).
Dolichol recycling
After the transfer of the oligosaccharide to nascent proteins, Dol-P-P is released in the lumenal leaflet of the ER (Fig.1). After one dephosphorylation step mediated by DOLPP1, Dol-P returns directly to the cytoplasmic leaflet where it can be re-used for LLO biosynthesis (Rush et al. 2008). The human DOLPP1 gene is the predicted ortholog of the CWH8/CAX4 yeast gene. The functional conservation through evolution was tested with the mouse gene Dolpp1 (Rush et al. 2002). In yeast, a defect in recycling Dol-P-P from the lumenal to the cytoplasmic leaflet of the ER impairs protein N-linked glycosylation (van Berkel et al. 1999; Fernandez et al. 2001). This indicates that this recycling of Dol-P-P contributes significantly to the pool of Dol-P available for glycosylation reactions.
Human disorders affecting the dolichol cycle
Phenotypes resulting from dolichol biosynthesis defects
Defects in the biosynthesis of N-glycans lead to diseases known as congenital disorders of glycosylation (CDG) (Freeze 2006; Jaeken and Matthijs 2007; Haeuptle and Hennet 2009). Research on CDG increases our understanding of consequences of a defect in the dolichol cycle. Recently, several diseases resulting from defects in dolichol biosynthesis (SRD5A3, DOLK) have been described, whereas other defects are closely associated with dolichol metabolism (MPDU1, DPM synthase). Unlike the disruption of genes strictly involved in the N-glycosylation process, defects in dolichol metabolism are predicted to affect several cellular pathways (Denecke and Kranz 2009).
Recently, the most upstream defect in the mevalonate pathway for dolichol-P synthesis was identified, it affects the reduction of polyprenol to dolichol. In a group of patients with variable eye malformations, cerebellar vermis hypoplasia, anemia, ichthyosiform dermatitis, liver dysfunction and coagulation abnormalities, we recently identified loss of function mutations in the SRD5A3 gene (SRD5A3-CDG; CDG-Iq) (Cantagrel et al. 2010; Morava et al. 2010). The 12 patients originally reported have an abnormal N-glycosylation profile of serum transferrin consistent with a role of polyprenol reductase in N-glycosylation. The transferrin isoelectric focusing shows a CDG type I profile with increased levels of asialo- and disialotransferrin. A family has also been reported without a clear diagnosis of CDG based on a routine transferrin test (Kahrizi et al. 2011). This observation might be the result of variabilities sometimes observed in CDG patients (Vermees et al. 2007) or might indicate that milder forms of this disorder can be missed with routine transferrin isoelectric focusing.
A deficiency one step further of dolichol kinase (MIM#610768; DOLK-CDG; CDG-Im) was described in four children from two families who died in their 1st year of life. Clinical features included dry ichthyosiform skin, seizures, severe muscular hypotonia, and progressive dilatative cardiomyopathy. The reported homozygous missense mutations almost abrogated the activity of the DOLK enzyme and were not able to complement the yeast dolichol kinase mutant (Kranz et al. 2007). Severe defects in N-glycosylation have been demonstrated using IEF of transferrin but some aspects of the phenotype might be caused by the involvement of other pathways. It can be speculated that less severe mutations in the DOLK gene can be responsible for a milder phenotype.
Dolichol-P is subsequently converted into dolichol-P-mannose for use as mannose donor in different ER glycosylation reactions. Mutations in the Dol-P-Man utilization protein (MPDU1), required for the availability of Dol-P-Man in the ER, lead to CDG-If. Four patients have been described (Schenck et al. 2001; Kranz et al. 2001) with mutations in the MPDU1 gene, presenting with early onset visual loss, seizures, psychomotor retardation and skin abnormalities like ichthyosyform skin. In all patients, N-glycosylation was found to be abnormal. Synthesis of Dol-P-Man is carried out by the DPM-synthase complex, consisting of DPM1-3 subunits. Mutations in DPM1 (CDG-Ie) were found in children with severe congenital visual loss, optic atrophy and seizures (Imbach et al. 2000; Kim et al. 2000), and one of the children was diagnosed with congenital frontal cortical hypoplasia (Garcia-Silva et al. 2004). Although no muscle histology was reported, all patients presented with hypotonia and increased CK. The relatively mild DPM3 defect was characterized by muscle dystrophy and dilated cardiomyopathy. Immunohistochemical analysis indicated a defect in alpha-dystroglycan O-mannosylation (Lefeber et al. 2009). No skin abnormalities were reported in the DPM synthase deficient patients.
Evidence for N-glycosylation defects
DOLK-CDG and SRD5A3-CDG have in common a clear defect in the ER part of the N-glycan biosynthesis (i.e., CDG type I). This defect is identified with the detection of glycoproteins with unoccupied N-glycosylation sites, both in patient serum and in the yeast models of these conditions. Multiple factors influence the variable site occupancy (Jones et al. 2005) including substrate concentrations. The pathophysiology of dolichol kinase and polyprenol reductase deficiencies show overlapping features with a likely effect on the Dol-P pool although it is less clear for SRD5A3 defects.
DOLK deficiency impacts directly the pool of dolichol phosphate, thereby limiting protein N-linked glycosylation and other glycosylation pathways via reduced Dol-P-Man formation. It is interesting to note that only a special pool out of the total dolichol phosphate present in the ER membrane is thought to participate actively in the glycosylation process (Hubbard and Robbins 1980; Gao and Lehrman 2002).
SRD5A3 deficiency may have an indirect effect on the dolichol phosphate pool. In vitro, dolichol phosphate is the limiting factor of LLO synthesis in cell extracts form SRD5A3 patient fibroblasts (Cantagrel et al. 2010). Although only a small percentage of dolichol is converted to Dol-P, SRD5A3 may contribute directly to the dolichol pool available for phosphorylation and LLO biosynthesis. With respect to the minimal amount of dolichol in the ER, further research into organelle specific effects of dolichol cycle defects will be required to obtain further insights. In the case of polyprenol reductase deficiency there is a clear accumulation of polyprenol detected in yeast, mouse and to a lesser extend in the serum of SRD5A3 patients. In these patients, serum levels of dolichols and polyprenols were elevated resulting in abnormal ratios of these lipids in favor of a block in the conversion of polyprenols to dolichol (Cantagrel et al. 2010; Morava et al. 2010). These serum polyisoprenoids are not available for protein glycosylation and they could result from regulation of cellular dolichol homeostasis. An intriguing question is if this accumulating precursor can have a deleterious effect on N-glycosylation or other cellular processes. In vitro studies indicate that sugar linked to polyprenol phosphate can be used by mammalian glycosyltransferases for LLO synthesis (D'Souza-Schorey et al. 1994), however, these substrates are not as effective as dolichol linked saccharides. It is not known if polyprenol can have an inhibitory effect on other dolichol-binding enzymes.
Evidence for O-mannosylation defects
O-mannosylation defects result in muscular dystrophy with elevated levels of creatine kinase and can be associated with varying degrees of cerebral cortex malformations, visual abnormalities and dilated cardiomyopathy. No muscular dystrophy has been detected in SRD5A3 patients and creatine kinase levels were normal when tested. Brain imaging revealed cerebellar vermis hypoplasia as the main feature in SRD5A3 patients whereas imaging of DOLK patients did not detect any malformations. A comparison with the MPDU1 and DPM disorders indicates that the muscular dystrophy and dilated cardiomyopathy could be shared between DOLK, DPM1 and DPM3, whereas these features are not present in SRD5A3 and MPDU1 defects.
For the neurological and visual problems, it is less clear how to correlate symptoms with pathophysiological pathways. Seizure are, for example, common in N-glycosylation and O-mannosylation defects, whereas deficiency of GPI anchor biosynthesis is associated with epilepsy as well (Krawitz et al. 2010). Ophthalmological involvement is known for both N-glycosylation and O-mannosylation defects.
Clinical features resulting from other defects
The presence of skin involvement in SRD5A3 and DOLK deficiencies might reflect a specific function for dolichol/dolichol phosphate in normal skin development. It is interesting to note that MPDU1-CDG and not DPM1 or DPM3-CDG shows similar skin involvement, a feature uncommon for N-glycosylation and O-mannosylation defects. This could suggest that the MPDU1 protein is involved in dolichol/dolichol-P homeostasis as well.
Future therapeutic options for dolichol biosynthesis defects
The in vitro rescue experiment of the LLO synthesis defect in cell extracts from SRD5A3 patients indicates that the adjustment of dolichol phosphate levels in patient cells could theoretically correct the N-glycosylation defect. Polyisoprenoid lipids are present in the diet, polyprenols are abundant in mature plants in contrast with animal tissues that possess dolichols. The contribution of dietary dolichol to the cellular pool of dolichol has been investigated in rat liver. An important part of the cholesterol utilized in metabolic processes is supplied by the diet but for polyisoprenoids, this contribution appeared to be quite limited. It was found that polyisoprenoids supplied orally were taken up through the rat gastrointestinal system; from 0.05% to a few percent of the total (Keller et al. 1982; Jakobsson et al. 1989). The uptake efficiency depends on the form of administration of dolichol and its incorporation into liposomes was found to be the most efficient. The study of the subcellular incorporation revealed that microsomes contain only a few percent of the labeled dietary dolichols. Making a significant part of this exogenous dolichol bioavailable will be an important step to consider some therapeutic options. Some previous studies detected that a fraction of dietary dolichol taken up by the liver was able to be phosphorylated (i.e., about 10% ) and potentially plays a role in protein glycosylation (Jakobsson et al. 1989) (Chojnacki and Dallner 1983).
Conclusion
Historically, mutations in genes encoding glycosyltransferases in the N-glycosylation process have been described as the cause of CDG. The identification of dolichol metabolism defects in patients with CDG opens a new field of metabolic diseases. They present unique opportunities to study the functional role of a poorly understood lipid, dolichol, in cellular processes. Besides its involvement in protein glycosylation via phosphorylation to dolichol-P in the ER, this will facilitate the identification of its role(s) in other organelles. Several steps involved in the synthesis, recycling and regulation of dolichol availability are still unknown. Exploration of these steps in humans might allow the identification of new inborn error of metabolisms in a spectrum overlapping partially with the Congenital Disorders of Glycosylation.
Reference
Acosta-Serrano A, O'Rear J et al. (2004) Defects in the N-linked oligosaccharide biosynthetic pathway in a Trypanosoma brucei glycosylation mutant. Eukaryot Cell 3(2):255–263
Behrens NH, Leloir LF (1970) Dolichol monophosphate glucose: an intermediate in glucose transfer in liver. Proc Natl Acad Sci USA 66(1):153–159
Belgareh-Touze N, Corral-Debrinski M et al. (2003) Yeast functional analysis: identification of two essential genes involved in ER to Golgi trafficking. Traffic 4(9):607–617
Cantagrel V, Lefeber DJ et al. (2010) SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142(2):203–217
Carson DD, Earles BJ, et al. (1981) Enhancement of protein glycosylation in tissue slices by dolichylphosphate. J Biol Chem 256(22):11552–11557
Chojnacki T, Dallner G (1983) The uptake of dietary polyprenols and their modification to active dolichols by the rat liver. J Biol Chem 258(2):916–922
Chojnacki T, Dallner G (1988) The biological role of dolichol. Biochem J 251(1):1–9
Denecke J, Kranz C (2009) Hypoglycosylation due to dolichol metabolism defects. Biochim Biophys Acta 1792(9):888–895
D'Souza-Schorey C, McLachlan KR et al. (1994) Mammalian glycosyltransferases prefer glycosyl phosphoryl dolichols rather than glycosyl phosphoryl polyprenols as substrates for oligosaccharyl synthesis. Arch Biochem Biophys 308(2):497–503
Edlund C, Soderberg M et al. (1994) Isoprenoids in aging and neurodegeneration. Neurochem Int 25(1):35–38
Ekstrom TJ, Chojnacki T et al. (1987) The alpha-saturation and terminal events in dolichol biosynthesis. J Biol Chem 262(9):4090–4097
Elmberger PG, Engfeldt P et al. (1988) Presence of dolichol and its derivatives in human blood. J Lipid Res 29(12):1651–1662
Ericsson J, Appelkvist EL et al. (1993) Biosynthesis of dolichol and cholesterol in rat liver peroxisomes. Biochimie 75(3–4):167–173
Fernandez F, Rush JS, et al. (2001) The CWH8 gene encodes a dolichyl pyrophosphate phosphatase with a luminally oriented active site in the endoplasmic reticulum of Saccharomyces cerevisiae. J Biol Chem 276(44):41455–41464
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7(7):537–551
Gao N, Lehrman MA, (2002) Coupling of the dolichol-P-P-oligosaccharide pathway to translation by perturbation-sensitive regulation of the initiating enzyme, GlcNAc-1-P transferase. J Biol Chem 277(42):39425–39435
Garcia-Silva MT, Matthijs G et al. (2004) Congenital disorder of glycosylation (CDG) type Ie. A new patient. J Inherit Metab Dis 27(5):591–600
Haeuptle MA, Hennet T (2009) Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 30(12):1628–1641
Heller L, Orlean P et al. (1992) Saccharomyces cerevisiae sec59 cells are deficient in dolichol kinase activity. Proc Natl Acad Sci USA 89(15):7013–7016
Hubbard SC, Robbins PW (1980) Synthesis of the N-linked oligosaccharides of glycoproteins. Assembly of the lipid-linked precursor oligosaccharide and its relation to protein synthesis in vivo. J Biol Chem 255(24):11782–11793
Imbach T, Schenk B et al. (2000) Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J Clin Invest 105(2):233–239
Jaeken J, Matthijs G (2007) Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 8:261–278
Jakobsson A, Swiezewska E et al. (1989) Uptake and modification of dietary polyprenols and dolichols in rat liver. FEBS Lett 255(1):32–36
Jones J, Krag SS et al. (2005) Controlling N-linked glycan site occupancy. Biochim Biophys Acta 1726(2):121–137
Jones MB, Rosenberg JN et al. (2009) Structure and synthesis of polyisoprenoids used in N-glycosylation across the three domains of life. Biochim Biophys Acta 1790(6):485–494
Kahrizi K, Hu CH et al. (2011) Next generation sequencing in a family with autosomal recessive Kahrizi syndrome (OMIM 612713) reveals a homozygous frameshift mutation in SRD5A3. Eur J Hum Genet 19(1):115–117
Keller RK, Jehle E et al. (1982) The origin of dolichol in the liver of the rat. Determination of the dietary contribution. J Biol Chem 257(15):8985–8989
Kim S, Westphal V et al. (2000) Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie). J Clin Invest 105(2):191–198
Krag SS (1998) The importance of being dolichol. Biochem Biophys Res Commun 243(1):1–5
Kranz C, Denecke J, et al. (2001) A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If). J Clin Invest 108(11):1613–1619
Kranz C, Jungeblut C, et al. (2007) A defect in dolichol phosphate biosynthesis causesa new inherited disorder with death in early infancy. Am J Hum Genet 80(3):433–440
Krawitz PM, Schweiger MR et al. (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42(10):827–829
Lefeber DJ, Schonberger J et al. (2009) Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 85(1):76–86
Morava E, Wevers RA et al. (2010) A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain 133(11):3210–3220
Pallottini V, Marino M et al. (2003) Age-related changes of isoprenoid biosynthesis in rat liver and brain. Biogerontology 4(6):371–378
Pennock JF, Hemming FW et al. (1960) Dolichol: a naturally occurring isoprenoid alcohol. Nature 186:470–472
Rip JW, Carroll KK (1985) Distribution, metabolism and excretion of [1-14C]dolichol injected intravenously into rats. Biochem J 227(3):705–710
Rip JW, Rupar CA et al. (1981) Localization of a dolichyl phosphate phosphatase in plasma membranes of rat liver. J Biol Chem 256(4):1929–1934
Rip JW, Rupar CA et al. (1985) Distribution, metabolism and function of dolichol and polyprenols. Prog Lipid Res 24(4):269–309
Rosenwald AG, Krag SS (1990) Lec9 CHO glycosylation mutants are defective in the synthesis of dolichol. J Lipid Res 31(3):523–533
Rosenwald AG, Stoll J et al. (1990) Regulation of glycosylation. Three enzymes compete for a common pool of dolichyl phosphate in vivo. J Biol Chem 265(24):14544–14553
Rush JS, Cho SK et al. (2002) Identification and characterization of a cDNA encoding a dolichyl pyrophosphate phosphatase located in the endoplasmic reticulum of mammalian cells. J Biol Chem 277(47):45226–45234
Rush JS, Gao N et al. (2008) Recycling of dolichyl monophosphate to the cytoplasmic leaflet of the endoplasmic reticulum after the cleavage of dolichyl pyrophosphate on the lumenal monolayer. J Biol Chem 283(7):4087–4093
Rush JS, Matveev S et al. (2010) Expression of functional bacterial undecaprenyl pyrophosphate synthase in the yeast rer2{Delta} mutant and CHO cells. Glycobiology 20(12):1585–1593
Sagami H, Kurisaki A, et al. (1993) Formation of dolichol from dehydrodolichol is catalyzed by NADPH-dependent reductase localized in microsomes of rat liver. J Biol Chem 268(14):10109–10113
Sato M, Sato K et al. (1999). The yeast RER2 gene, identified by endoplasmic reticulum protein localization mutations, encodes cis-prenyltransferase, a key enzyme in dolichol synthesis. Mol Cell Biol 19(1):471–483.
Schenk B, Fernandez F et al. (2001) The ins(ide) and out(side) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology 11(5):61R–70R
Shridas P, Waechter CJ (2006) Human dolichol kinase, a polytopic endoplasmic reticulum membrane protein with a cytoplasmically oriented CTP-binding site. J Biol Chem 281(42):31696–31704
Shridas P, Rush JS et al. (2003) Identification and characterization of a cDNA encoding a long-chain cis-isoprenyltranferase involved in dolichyl monophosphate biosynthesis in the ER of brain cells. Biochem Biophys Res Commun 312(4):1349–1356
Skorupinska-Tudek K, Wojcik J et al. (2008) Polyisoprenoid alcohols–recent results of structural studies. Chem Rec 8(1):33–45
Spiro MJ, Spiro RG (1986) Control of N-linked carbohydrate unit synthesis in thyroid endoplasmic reticulum by membrane organization and dolichyl phosphate availability. J Biol Chem 261(31):14725–14732
Swiezewska E, Danikiewicz W (2005) Polyisoprenoids: structure, biosynthesis and function. Prog Lipid Res 44(4):235–258
Takahashi S, Koyama T (2006) Structure and function of cis-prenyl chain elongating enzymes. Chem Rec 6(4):194–205
Tollbom O, Dallner G (1986) Dolichol and dolichyl phosphate in human tissues. Br J Exp Pathol 67(5):757–764
Turner DC, Gruner SM (1992) X-ray diffraction reconstruction of the inverted hexagonal (HII) phase in lipid-water systems. Biochemistry 31(5):1340–1355
Turunen M, Schedin-Weiss S (2007) Defect in fatty acid esterification of dolichol in Niemann-Pick type C1 mouse livers in vivo. Biochim Biophys Acta 1771(4):506–513
Valtersson C, Van Duyn G et al. (1985) The influence of dolichol, dolichol esters, and dolichyl phosphate on phospholipid polymorphism and fluidity in model membranes. J Biol Chem 260(5):2742–2751
van Berkel MA, Rieger M, et al. (1999) The Saccharomyces cerevisiae CWH8 gene is required for full levels of dolichol-linked oligosaccharides in the endoplasmic reticulum and for efficient N-glycosylation. Glycobiology 9(3):243–253
Van Duijn G, Valtersson C et al. (1986) Dolichyl phosphate induces non-bilayer structures, vesicle fusion and transbilayer movement of lipids: a model membrane study. Biochim Biophys Acta 861(2):211–223
Van Houte HA, Van Veldhoven PP et al. (1997) Metabolism of dolichol, dolichoic acid and nordolichoic acid in cultured cells. Biochim Biophys Acta 1347(1):93–100
Vermeer S, Kremer HP et al. (2007) Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol 254(10):1356–1358
Ward WC, Guan Z et al. (2007) Identification and quantification of dolichol and dolichoic acid in neuromelanin from substantia nigra of the human brain. J Lipid Res 48(7):1457–1462
Wolf MJ, Rush JS et al. (1991) Golgi-enriched membrane fractions from rat brain and liver contain long-chain polyisoprenyl pyrophosphate phosphatase activity. Glycobiology 1(4):405–410
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by: Jaak Jaeken
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cantagrel, V., Lefeber, D.J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J Inherit Metab Dis 34, 859–867 (2011). https://doi.org/10.1007/s10545-011-9301-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9301-0