
Abstract
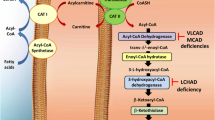
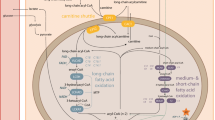
Mouse models have been designed for a number of fatty acid oxidation defects. Studies in these mouse models have demonstrated that different pathogenetic mechanisms play a role in the pathophysiology of defects of fatty acid oxidation. Supplementation with L-carnitine does not prevent low tissue carnitine levels and induces acylcarnitine production having potentially toxic effects, as presented in very-long-chain acyl-CoA dehydrogenase (VLCAD)-deficient mice. Energy deficiency appears to be an important mechanism in the development of cardiomyopathy and skeletal myopathy in fatty acid oxidation defects and is also the underlying mechanism of cold intolerance. Hypoglycemia as one major clinical sign in all fatty acid oxidation defects occurs due to a reduced hepatic glucose output and an enhanced peripheral glucose uptake rather than to transcriptional changes that are also observed simultaneously, as presented in medium-chain acyl-CoA dehydrogenase (MCAD)-deficient mice. There are reports that an impaired fatty acid oxidation also plays a role in intrauterine life. The embryonic loss demonstrated for some enzyme defects in the mouse supports this hypothesis. However, the exact mechanisms are unknown. This observation correlates to maternal hemolysis, elevated liver enzymes, low platelets (HELLP) syndrome, as observed in pregnancies carrying a long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD)-deficient fetus. Synergistic heterozygosity has been shown in isolated patients and in mouse models to be associated with clinical phenotypes common to fatty acid oxidation disorders. Synergistic mutations may also modulate severity of the clinical phenotype and explain in part clinical heterogeneity of fatty acid oxidation defects. In summary, knowledge about the different pathogenetic mechanisms and the resulting pathophysiology allows the development of specific new therapies.
Similar content being viewed by others

Abbreviations
- APR:
-
acute-phase response
- CPT-1a:
-
carnitine palmitoyl-CoA transferase-1a (liver isoform)
- CPT-1b:
-
carnitine palmitoyl-CoA transferase-1b (muscle isoform)
- DAG:
-
diacylglycerol
- FAO:
-
fatty acid oxidation
- G-6-P:
-
glucose-6 phosphate
- LCAD:
-
long-chain acyl-CoA dehydrogenase
- LCT:
-
long-chain triglycerides
- MCAD:
-
medium-chain acyl-CoA dehydrogenase
- MCADD:
-
medium-chain acyl-CoA dehydrogenase deficiency
- MCT:
-
medium-chain triglycerides
- PPAR-α:
-
peroxisomal proliferator activated receptor alpha
- PGC1-α:
-
PPAR-γ coactivator-1 alpha
- PDK4:
-
pyruvate dehydrogenase kinase 4
- SCAD:
-
short-chain acyl-CoA dehydrogenase
- TFP:
-
mitochondrial trifunctional protein
- TFPD:
-
trifunctional protein deficiency
- VLCAD:
-
very-long-chain acyl-CoA dehydrogenase
- VLCADD:
-
very-long-chain acyl-CoA dehydrogenase deficiency
References
Amendt BA, Freneaux E, Reece C, Wood PA, Rhead WJ (1992) Short-chain acyl-coenzyme A dehydrogenase activity, antigen, and biosynthesis are absent in the BALB/cByJ mouse. Pediatr Res 31:552–556
Armstrong DL, Masiowski ML, Wood PA (1993) Pathologic characterization of short-chain acyl-CoA dehydrogenase deficiency in BALB/cByJ mice. Am J Med Genet 47:884–892
Berger PS, Wood PA (2004) Disrupted blastocoele formation reveals a critical developmental role for long-chain acyl-CoA dehydrogenase. Mol Genet Metab 82:266–272
Bonnet D, Martin D, de Lonlay P et al (1999) Arrhythmias and conduction defects as presenting symptoms in fatty acid oxidation disorders in children. Circulation 100:2248–2253
Brown-Harrison MC, Nada MA, Sprecher H et al (1996) Very long chain acyl-CoA dehydrogenase deficiency: successful treatment of acute cardiomyopathy. Biochem Mol Med 58:59–65
Cox KB, Hamm DA, Millington DS et al (2001) Gestational, pathologic, and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum Mol Genet 10:2069–2077
Cox KB, Liu J, Tian L, Barnes S, Yang Q, Wood PA (2009) Cardiac hypertrophy in mice with long-chain acyl-CoA dehydrogenase or very long-chain acyl-CoA dehydrogenase deficiency. Lab Invest 89(12):1348–1354
Enerback S, Jacobsson A, Simpson EM et al (1997) Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387:90–94
Exil VJ, Roberts RL, Sims H et al (2003) Very-long-chain Acyl-coenzyme A dehydrogenase deficiency in mice. Circ Res 93:448–455
Exil VJ, Gardner CD, Rottman JN et al (2006) Abnormal mitochondrial bioenergetics and heart rate dysfunction in mice lacking very-long-chain acyl-CoA dehydrogenase. Am J Physiol Heart Circ Physiol 290:H1289–H1297
Gillingham MB, Scott B, Elliott D, Harding CO (2006) Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab 89:58–63
Goetzman ES, Tian L, Wood PA (2005) Differential induction of genes in liver and brown adipose tissue regulated by peroxisome-proliferator activated receptor-alpha during fasting and cold exposure in acyl-CoA dehydrogenase-deficient mice. Mol Genet Metab 84:39–47
Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP (1998) Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. J Clin Invest 102:1724–1731
Herrema H, Derks TG, van Dijk TH et al (2008) Disturbed hepatic carbohydrate management during high metabolic demand in medium-chain acyl-CoA dehydrogenase (MCAD)-deficient mice. Hepatology 47:1894–1904
Hinsdale ME, Kelly CL, Wood PA (1993) Null allele at Bcd-1 locus in BALB/cByJ mice is due to a deletion in the short-chain acyl-CoA dehydrogenase gene and results in missplicing of mRNA. Genomics 16:605–611
Ibdah JA, Paul H, Zhao Y, Binford S et al (2001) Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest 107:1403–1409
Ji S, You Y, Kerner J et al (2008) Homozygous carnitine palmitoyltransferase 1b (muscle isoform) deficiency is lethal in the mouse. Mol Genet Metab 93:314–322
Kurtz DM, Rinaldo P, Rhead WJ et al (1998) Targeted disruption of mouse long-chain acyl-CoA dehydrogenase reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci USA 95:15592–15597
Nyman LR, Cox KB, Hoppel CL et al (2005) Homozogous carnitine palmitoyltransferase 1a (liver isoform) deficiency is lethal in the mouse. Mol Genet Metab 86:179–187
Oey NA, Ruiter JP, Attié-Bitach T, Ijlst L, Wanders RJ, Wijburg FA (2006) Fatty acid oxidation in the human fetus: implications for fetal and adult disease. J Inherit Metab Dis 29:71–75
Primassin S, Ter Veld F, Mayatepek E, Spiekerkoetter U (2008) Carnitine supplementation induces acylcarnitine production in tissues of very long-chain acyl-CoA dehydrogenase-deficient mice, without replenishing low free carnitine. Pediatr Res 63:632–637
Rakheja D, Bennett MJ, Foster BM, Domiati-Saad R, Rogers BB (2002) Evidence for fatty acid oxidation in human placenta, and the relationship of fatty acid oxidation enzyme activities with gestational age. Placenta 23:447–450
Schuler AM, Wood PA (2002) Mouse models for disorders of mitochondrial fatty acid beta-oxidation. ILAR J 43:57–65
Schuler AM, Gower BA, Matern D, Rinaldo P, Vockley J, Wood PA (2005) Synergistic heterozygosity in mice with inherited enzyme deficiencies of mitochondrial fatty acid β-oxidation. Mol Genet Metab 85:7–11
Schymik I, Liebig M, Mueller M et al (2006) Pitfalls of neonatal screening for very-long-chain acyl-CoA dehydrogenase deficiency using tandem mass spectrometry. J Pediatr 149:128–130
Spiekerkoetter U (2007) Effects of a fat load and exercise on asymptomatic VLCAD deficiency. J Inherit Metab Dis 30:405
Spiekerkoetter U, Tenenbaum T, Heusch A, Wendel U (2003) Cardiomyopathy and pericardial effusion in infancy point to a fatty acid beta-oxidation defect after exclusion of an underlying infection. Pediatr Cardiol 24:295–297
Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW (2004a) General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res 55:190–196
Spiekerkoetter U, Tokunaga C, Wendel U et al (2004b) Changes in blood carnitine and acylcarnitine profiles of very long-chain acyl-CoA dehydrogenase-deficient mice subjected to stress. Eur J Clin Invest 34:191–196
Spiekerkoetter U, Tokunaga C, Wendel U et al (2005) Tissue carnitine homeostasis in very-long-chain acyl-CoA dehydrogenase-deficient mice. Pediatr Res 57:760–764
Spiekerkoetter U, Mueller M, Cloppenburg E et al (2008) Intrauterine cardiomyopathy and cardiac mitochondrial proliferation in mitochondrial trifunctional protein (TFP) deficiency. Mol Genet Metab 94:428–430
Spiekerkoetter U, Lindner M, Santer R et al (2009) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32:488–497
ter Veld F, Jacoby C, Floegel U, Spiekerkoetter U. (2007) Impaired cardiac function in very long-chain acyl-CoA dehydrogenase-deficient mice as studied by in-vivo NMR. JIMD 30: 201-P
ter Veld F, Primassin S, Hoffmann L, Mayatepek E, Spiekerkoetter U (2009) Corresponding increase in long-chain acyl-CoA and acylcarnitine after exercise in muscle from VLCAD mice. J Lipid Res 50:1556–1562
Tolwani RJ, Hamm DA, Tian L, et al. (2005) Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet. 1(2):e23. Epub 2005 Aug 19.
van Vlies N, Tian L, Overmars H et al (2005) Characterization of carnitine and fatty acid metabolism in the long-chain acyl-CoA dehydrogenase-deficient mouse. Biochem J 387:185–193
Vaz FM, Wanders RJ (2002) Carnitine biosynthesis in mammals. Biochem J 361:417–429
Vockley J, Rinaldo P, Bennett M, Matern D, Vladutiu G (2000) Synergistic Heterozygosity: Disease resulting from multiple partial defects in one or more metabolic pathways. Mol Genet Metab 71:10–18
Wanders RJA, Vreken P, Den Boer MEJ et al (1999) Disorders of mitochondrial fatty acyl-CoA β-oxidation. J Inherit Metab Dis 22:442–487
Winter SC (2003) Treatment of carnitine deficiency. J Inherit Metab Dis. 26:171–180
Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D (1989) Short-chain acyl-coenzyme A dehydrogenase deficiency in mice. Pediatr Res 25:38–43
Acknowledgements
Part of the work supported by grants from the Deutsche Forschungsgemeinschaft to US (DFG, SP1125/1-1; SFB 575; SFB 612). Other parts of the work were supported by National Institutes of Health (NIH) Grant Number RO1-RR02599 and T-32-RR00493 to PAW from the National Center for Research Resources (NCRR), a component of the NIH, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Verena Peters
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Spiekerkoetter, U., Wood, P.A. Mitochondrial fatty acid oxidation disorders: pathophysiological studies in mouse models. J Inherit Metab Dis 33, 539–546 (2010). https://doi.org/10.1007/s10545-010-9121-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9121-7